
Key Points
-
Transmissible spongiform encephalopathies (TSEs) are neurodegenerative diseases of humans and many animal species that are caused by prions. The main constituent of prions is scrapie prion protein (PrPSc), an aggregated moiety of the host-derived membrane glycolipoprotein cellular prion protein (PrPC). Although PrPC is encoded by the host genome, prions were found to encipher many phenotypic TSE variants, known as prion strains.
-
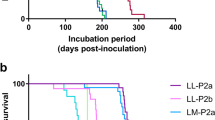
Prion strains are TSE isolates that, when inoculated into new hosts, consistently cause disease with specific characteristics, such as incubation period, patterns of PrPSc distribution and relative severity of spongiform changes in the brain (the lesion profile).The agent-specified information of prion strains is thought to be contained within distinct conformations of various PrPSc isotypes.
-
Prions exert their destructive effects predominantly, if not exclusively, within the central nervous system. However, the direct cause of neurotoxicity remains unclear. PrPC is required for prion replication because mice that lack PrPC are resistant to prions. The presence of PrPC on neurons is a prerequisite for prion-induced neurotoxicity.
-
A series of transgenic mice that express various prion protein mutants indicate that deletion of specific regions of PrPC can render it neurotoxic. This toxicity is modulated by co-expression of wild-type PrPC.
-
Currently, there is no reagent allowing non-invasive, pre-mortem diagnosis of prion diseases. In view of recent unfortunate cases of Creutzfeldt–Jakob disease infection through blood transfusion, reliable, specific and, most importantly, sensitive reagents are urgently needed.
Abstract
Transmissible spongiform encephalopathies (TSEs) are neurodegenerative diseases that are caused by prions and affect humans and many animal species. It is now widely accepted that the infectious agent that causes TSEs is PrPSc, an aggregated moiety of the host-derived membrane glycolipoprotein PrPC. Although PrPC is encoded by the host genome, prions themselves encipher many phenotypic TSE variants, known as prion strains. Prion strains are TSE isolates that, after inoculation into distinct hosts, cause disease with consistent characteristics, such as incubation period, distinct patterns of PrPSc distribution and spongiosis and relative severity of the spongiform changes in the brain. The existence of such strains poses a fascinating challenge to prion research.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Glatzel, M. et al. Human prion diseases: epidemiology and integrated risk assessment. Lancet Neurol. 2, 757–763 (2003).
Collins, P. S., Lawson, V. A. & Masters, P. C. Transmissible spongiform encephalopathies. Lancet 363, 51–61 (2004).
Prusiner, S. B. Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 (1982). Enunciation of the 'protein-only' hypothesis and definition of the prion.
Griffith, J. S. Self-replication and scrapie. Nature 215, 1043–1044 (1967). First communication of the 'protein-only' hypothesis.
Aguzzi, A. & Heikenwalder, M. Prion diseases: cannibals and garbage piles. Nature 423, 127–129 (2003).
Oesch, B., Groth, D. F., Prusiner, S. B. & Weissmann, C. Search for a scrapie-specific nucleic acid: a progress report. Ciba Found. Symp. 135, 209–223 (1988).
Oesch, B. et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 40, 735–746 (1985).
Prusiner, S. B. Creutzfeldt-Jakob disease and scrapie prions. Alzheimer Dis. Assoc. Disord. 3, 52–78 (1989).
Weissmann, C. A 'unified theory' of prion propagation. Nature 352, 679–683 (1991).
Büeler, H. R. et al. Mice devoid of PrP are resistant to scrapie. Cell 73, 1339–1347 (1993). Demonstration that Prnp0/0 mice are resistant to prion disease.
Aguzzi, A. & Polymenidou, M. Mammalian prion biology. One century of evolving concepts. Cell 116, 313–327 (2004).
Zhang, C. C., Steele, A. D., Lindquist, S. & Lodish, H. F. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc. Natl Acad. Sci. USA 103, 2184–2189 (2006).
Steele, A. D., Emsley, J. G., Ozdinler, P. H., Lindquist, S. & Macklis, J. D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl Acad. Sci. USA 103, 3416–3421 (2006).
Silveira, J. R. et al. The most infectious prion protein particles. Nature 437, 257–261 (2005).
Telling, G. C. et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83, 79–90 (1995).
Priola, S. A., Chesebro, B. & Caughey, B. Biomedicine. A view from the top – prion diseases from 10,000 feet. Science 300, 917–919 (2003).
Bolton, D. C., McKinley, M. P. & Prusiner, S. B. Identification of a protein that purifies with the scrapie prion. Science 218, 1309–1311 (1982).
Safar, J. G. et al. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nature Biotech. 20, 1147–1150 (2002).
Sigurdson, C. J. et al. Strain fidelity of chronic wasting disease upon murine adaptation. J. Virol. 80, 12303–12311 (2006).
Richt, J. A. et al. Characterization of the recent U.S. BSE case and methods for surveillance. Proc. Annu. Meet. US Anim. Health Assoc. 108, 91–92 (2004).
Heikenwalder, M. et al. Chronic lymphocytic inflammation specifies the organ tropism of prions. Science 307, 1107–1110 (2005). Inflammatory conditions support prion replication in otherwise prion-free organs.
Seeger, H. et al. Coincident scrapie infection and nephritis lead to urinary prion excretion. Science 310, 324–326 (2005).
Ligios, C. et al. PrPSc in mammary glands of sheep affected by scrapie and mastitis. Nature Med. 11, 1137–1138 (2005).
Houston, F., Foster, J. D., Chong, A., Hunter, N. & Bostock, C. J. Transmission of BSE by blood transfusion in sheep. Lancet 356, 999–1000 (2000).
Mathiason, C. K. et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 314, 133–136 (2006).
Llewelyn, C. A. et al. Possible transmission of variant Creutzfeldt–Jakob disease by blood transfusion. Lancet 363, 417–421 (2004).
Peden, A. H., Head, M. W., Ritchie, D. L., Bell, J. E. & Ironside, J. W. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364, 527–529 (2004). A case report of accidental CJD transmission through blood transfusion.
Wroe, S. J. et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt–Jakob disease associated with blood transfusion: a case report. Lancet 368, 2061–2067 (2006).
Aguzzi, A. & Glatzel, M. Prion infections, blood and transfusions. Nature Clin. Pract. Neurol. 2, 321–329 (2006).
Safar, J. et al. Eight prion strains have PrPSc molecules with different conformations. Nature Med. 4, 1157–1165 (1998).
Nonno, R. et al. Efficient transmission and characterization of Creutzfeldt–Jakob disease strains in bank voles. PLoS Pathog. 2, e12 (2006).
Shorter, J. & Lindquist, S. Prions as adaptive conduits of memory and inheritance. Nature Rev. Genet. 6, 435–450 (2005).
Wickner, R. B., Edskes, H. K., Roberts, B. T., Pierce, M. & Baxa, U. Prions of yeast as epigenetic phenomena: high protein 'copy number' inducing protein 'silencing'. Adv. Genet. 46, 485–525 (2002).
Aguzzi, A. Understanding the diversity of prions. Nature Cell Biol. 6, 290–292 (2004).
Pattison, I. H. & Millson, G. C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. 71, 101–108 (1961).
Fraser, H. & Dickinson, A. G. Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J. Comp. Pathol. 83, 29–40 (1973).
Bruce, M. E. & Dickinson, A. G. Biological evidence that scrapie agent has an independent genome. J. Gen. Virol. 68, 79–89 (1987).
Kimberlin, R. H., Cole, S. & Walker, C. A. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J. Gen. Virol. 68, 1875–1881 (1987).
Race, R. et al. Subclinical scrapie infection in a resistant species: persistence, replication, and adaptation of infectivity during four passages. J. Infect. Dis. 186 (Suppl. 2), S166–S170 (2002).
Hill, A. F. & Collinge, J. Subclinical prion infection in humans and animals. Br. Med. Bull. 66, 161–170 (2003).
Hill, A. F. et al. Species-barrier-independent prion replication in apparently resistant species. Proc. Natl Acad. Sci. USA 29, 10248–10253 (2000).
Collinge, J. et al. Unaltered susceptibility to BSE in transgenic mice expressing human prion protein. Nature 378, 779–783 (1995).
Scott, M. R. et al. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc. Natl Acad. Sci. USA 96, 15137–151342 (1999).
Scott, M. R., Peretz, D., Nguyen, H. O., Dearmond, S. J. & Prusiner, S. B. Transmission barriers for bovine, ovine, and human prions in transgenic mice. J. Virol. 79, 5259–5271 (2005).
Scott, M. et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59, 847–857 (1989).
Vilotte, J. L. et al. Markedly increased susceptibility to natural sheep scrapie of transgenic mice expressing ovine PrP. J. Virol. 75, 5977–5984 (2001).
Collinge, J., Sidle, K. C., Meads, J., Ironside, J. & Hill, A. F. Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD. Nature 383, 685–690 (1996).
Collinge, J. Molecular neurology of prion disease. J. Neurol. Neurosurg. Psychiatr. 76, 906–919 (2005).
Khalili-Shirazi, A. et al. PrP glycoforms are associated in a strain-specific ratio in native PrPSc. J. Gen. Virol. 86, 2635–2644 (2005).
DeArmond, S. J. et al. Selective neuronal targeting in prion disease. Neuron 19, 1337–1348 (1997).
Neuendorf, E. et al. Glycosylation deficiency at either one of the two glycan attachment sites of cellular prion protein preserves susceptibility to bovine spongiform encephalopathy and scrapie infections. J. Biol. Chem. 279, 53306–53316 (2004).
Cancellotti, E. et al. Altered glycosylated PrP proteins can have different neuronal trafficking in brain but do not acquire scrapie-like properties. J. Biol. Chem. 280, 42909–42918 (2005).
Bessen, R. A. & Marsh, R. F. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 68, 7859–7868 (1994). Describes the differentiation of prion strains by proteolytic digest.
Bessen, R. A. et al. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 375, 698–700 (1995).
Telling, G. C. et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274, 2079–2082 (1996).
Parchi, P. et al. Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl Acad. Sci. USA 97, 10168–10172 (2000).
Zanusso, G. et al. Identification of distinct N-terminal truncated forms of prion protein in different Creutzfeldt–Jakob disease subtypes. J. Biol. Chem. 279, 38936–38942 (2004).
Bruce, M. E. et al. Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature 389, 498–501 (1997).
Palmer, M. S., Dryden, A. J., Hughes, J. T. & Collinge, J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt–Jakob disease. Nature 352, 340–342 (1991).
Collinge, J., Palmer, M. S. & Dryden, A. J. Genetic predisposition to iatrogenic Creutzfeldt–Jakob disease. Lancet 337, 1441–1442 (1991).
Puoti, G. et al. Sporadic Creutzfeldt–Jakob disease: co-occurrence of different types of PrPSc in the same brain. Neurology 53, 2173–2176 (1999).
Dickson, D. W. & Brown, P. Multiple prion types in the same brain: is a molecular diagnosis of CJD possible?. Neurology 53, 1903–1904 (1999).
Polymenidou, M. et al. Coexistence of multiple PrPSc types in individuals with Creutzfeldt–Jakob disease. Lancet Neurol. 4, 805–814 (2005).
Yull, H. M. et al. Detection of type 1 prion protein in variant Creutzfeldt–Jakob disease. Am. J. Pathol. 168, 151–157 (2006).
Nishida, N., Katamine, S. & Manuelidis, L. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science 310, 493–496 (2005).
Bartz, J. C. et al. Prion interference is due to a reduction in strain-specific PrPSc levels. J. Virol. 81, 689–697 (2007).
Dickinson, A. G., Fraser, H., Meikle, V. M. & Outram, G. W. Competition between different scrapie agents in mice. Nature New Biol. 237, 244–245 (1972).
Manuelidis, L. Vaccination with an attenuated Creutzfeldt–Jakob disease strain prevents expression of a virulent agent. Proc. Natl Acad. Sci. USA 95, 2520–2525 (1998).
Casalone, C. et al. Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt–Jakob disease. Proc. Natl Acad. Sci. USA 101, 3065–3070 (2004).
Beringue, V. et al. Isolation from cattle of a prion strain distinct from that causing bovine spongiform encephalopathy. PLoS Pathog. 2, e112 (2006).
Meyer-Luehmann, M. et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 313, 1781–1784 (2006).
Aguzzi, A. & Haass, C. Games played by rogue proteins in prion disorders and Alzheimer's disease. Science 302, 814–818 (2003).
Riek, R. Cell biology: infectious Alzheimer's disease? Nature 444, 429–431 (2006).
Aguzzi, A. & Sigurdson, C. J. Antiprion immunotherapy: to suppress or to stimulate? Nature Rev. Immunol. 4, 725–736 (2004).
Brown, P. et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann. Neurol. 35, 513–529 (1994).
Glatzel, M., Abela, E., Maissen, M. & Aguzzi, A. Extraneural pathologic prion protein in sporadic Creutzfeldt–Jakob disease. N. Engl. J. Med. 349, 1812–1820 (2003).
Peden, A. H., Ritchie, D. L., Head, M. W. & Ironside, J. W. Detection and localization of PrPSc in the skeletal muscle of patients with variant, iatrogenic, and sporadic forms of Creutzfeldt–Jakob disease. Am. J. Pathol. 168, 927–935 (2006).
Blättler, T. et al. PrP-expressing tissue required for transfer of scrapie infectivity from spleen to brain. Nature 389, 69–73 (1997).
Brown, K. L. et al. Scrapie replication in lymphoid tissues depends on prion protein- expressing follicular dendritic cells. Nature Med. 5, 1308–1312 (1999).
Bartz, J. C., Dejoia, C., Tucker, T., Kincaid, A. E. & Bessen, R. A. Extraneural prion neuroinvasion without lymphoreticular system infection. J. Virol. 79, 11858–11863 (2005).
Bessen, R. A. & Marsh, R. F. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 66, 2096–2101 (1992).
Telling, G. C. et al. Transmission of Creutzfeldt–Jakob disease from humans to transgenic mice expressing chimeric human–mouse prion protein. Proc. Natl Acad. Sci. USA 91, 9936–9940 (1994).
Büeler, H. R. et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582 (1992).
Mallucci, G. R. et al. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 21, 202–210 (2002).
Meier, P. et al. Soluble dimeric prion protein binds PrPScin vivo and antagonizes prion disease. Cell 113, 49–60 (2003).
Brandner, S. et al. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379, 339–343 (1996).
Mallucci, G. et al. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302, 871–874 (2003). Describes a role for PrPC in prion-induced neurotoxicity.
Chesebro, B. et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308, 1435–1439 (2005).
Aguzzi, A. Cell biology. Prion toxicity: all sail and no anchor. Science 308, 1420–1421 (2005).
Kimberley, F. C., Sivasankar, B. & Paul Morgan, B. Alternative roles for CD59. Mol. Immunol. 44, 73–81 (2007).
Trifilo, M. J. et al. Prion-induced amyloid heart disease with high blood infectivity in transgenic mice. Science 313, 94–97 (2006).
Shmerling, D. et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93, 203–214 (1998).
Radovanovic, I. et al. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J. Neurosci. 25, 4879–4888 (2005).
Baumann, F. et al. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 26, 538–547 (2007).
Hegde, R. S. et al. A transmembrane form of the prion protein in neurodegenerative disease. Science 279, 827–834 (1998).
Hegde, R. S. et al. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature 402, 822–826 (1999).
Stewart, R. S., Piccardo, P., Ghetti, B. & Harris, D. A. Neurodegenerative illness in transgenic mice expressing a transmembrane form of the prion protein. J. Neurosci. 25, 3469–3477 (2005).
Ma, J., Wollmann, R. & Lindquist, S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298, 1781–1785 (2002).
Ma, J. & Lindquist, S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science 298, 1785–1788 (2002).
Drisaldi, B. et al. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J. Biol. Chem. 278, 21732–21743 (2003).
Roucou, X., Guo, Q., Zhang, Y., Goodyer, C. G. & LeBlanc, A. C. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J. Biol. Chem. 278, 40877–40881 (2003).
Barmada, S., Piccardo, P., Yamaguchi, K., Ghetti, B. & Harris, D. A. GFP-tagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol. Dis. 16, 527–537 (2004).
Barmada, S. J. & Harris, D. A. Visualization of prion infection in transgenic mice expressing green fluorescent protein-tagged prion protein. J. Neurosci. 25, 5824–5832 (2005).
Ford, M. J., Burton, L. J., Morris, R. J. & Hall, S. M. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience 113, 177–192 (2002).
Herms, J. et al. Evidence of presynaptic location and function of the prion protein. J. Neurosci. 19, 8866–8875 (1999).
Moser, M., Colello, R. J., Pott, U. & Oesch, B. Developmental expression of the prion protein gene in glial cells. Neuron 14, 509–517 (1995).
Stahl, N., Borchelt, D. R., Hsiao, K. & Prusiner, S. B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51, 229–240 (1987).
Haraguchi, T. et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 274, 1–13 (1989).
Riek, R. et al. NMR structure of the mouse prion protein domain PrP (121–321). Nature 382, 180–182 (1996).
Wuthrich, K. & Riek, R. Three-dimensional structures of prion proteins. Adv. Protein Chem. 57, 55–82 (2001).
Knaus, K. J. et al. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nature Struct. Biol. 8, 770–774 (2001).
Hornemann, S. et al. Recombinant full-length murine prion protein, mPrP (23–231): purification and spectroscopic characterization. FEBS Lett. 413, 277–281 (1997).
Brown, D. R. et al. The cellular prion protein binds copper in vivo. Nature 390, 684–687 (1997).
Garnett, A. P. & Viles, J. H. Copper binding to the octarepeats of the prion protein. Affinity, specificity, folding, and cooperativity: insights from circular dichroism. J. Biol. Chem. 278, 6795–6802 (2003).
Jones, C. E., Klewpatinond, M., Abdelraheim, S. R., Brown, D. R. & Viles, J. H. Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: the unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations. J. Mol. Biol. 346, 1393–1407 (2005).
Brown, D. R., Schulz-Schaeffer, W. J., Schmidt, B. & Kretzschmar, H. A. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 146, 104–112 (1997).
Waggoner, D. J. et al. Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J. Biol. Chem. 275, 7455–7458 (2000).
Hutter, G., Heppner, F. L. & Aguzzi, A. No superoxide dismutase activity of cellular prion protein in vivo. Biol. Chem. 384, 1279–1285 (2003).
Enari, M., Flechsig, E. & Weissmann, C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl Acad. Sci. USA 98, 9295–9299 (2001).
Peretz, D. et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412, 739–743 (2001).
Heppner, F. L. et al. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 294, 178–182 (2001).
White, A. R. et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 422, 80–83 (2003).
Koller, M. F., Grau, T. & Christen, P. Induction of antibodies against murine full-length prion protein in wild-type mice. J. Neuroimmunol. 132, 113–116 (2002).
Sigurdsson, E. M. et al. Immunization delays the onset of prion disease in mice. Am. J. Pathol. 161, 13–17 (2002).
Sigurdsson, E. M. et al. Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci. Lett. 336, 185–187 (2003).
Tal, Y. et al. Complete Freund's adjuvant immunization prolongs survival in experimental prion disease in mice. J. Neurosci. Res. 71, 286–290 (2003).
Souan, L., Margalit, R., Brenner, O., Cohen, I. R. & Mor, F. Self prion protein peptides are immunogenic in Lewis rats. J. Autoimmun. 17, 303–310 (2001).
Souan, L. et al. Modulation of proteinase-K resistant prion protein by prion peptide immunization. Eur. J. Immunol. 31, 2338–2346 (2001).
Schwarz, A. et al. Immunisation with a synthetic prion protein-derived peptide prolongs survival times of mice orally exposed to the scrapie agent. Neurosci. Lett. 350, 187–189 (2003).
Arbel, M., Lavie, V. & Solomon, B. Generation of antibodies against prion protein in wild-type mice via helix 1 peptide immunization. J. Neuroimmunol. 144, 38–45 (2003).
Gregoire, S. et al. Identification of two immunogenic domains of the prion protein — PrP — which activate class II-restricted T cells and elicit antibody responses against the native molecule. J. Leukoc. Biol. 76, 125–134 (2004).
Rosset, M. B. et al. Breaking immune tolerance to the prion protein using prion protein peptides plus oligodeoxynucleotide-CpG in mice. J. Immunol. 172, 5168–5174 (2004).
Polymenidou, M. et al. Humoral immune response to native eukaryotic prion protein correlates with anti-prion protection. Proc. Natl Acad. Sci. USA 101, 14670–14676 (2004).
Lechner, F. et al. Virus-like particles as a modular system for novel vaccines. Intervirology 45, 212–217 (2002).
Nikles, D. et al. Circumventing tolerance to the prion protein (PrP): vaccination with PrP-displaying retrovirus particles induces humoral immune responses against the native form of cellular PrP. J. Virol. 79, 4033–4042 (2005).
Solforosi, L. et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303, 1514–1516 (2004).
Nicoll, J. A. et al. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nature Med. 9, 448–452 (2003).
Acknowledgements
A.A. is supported by grants from the European Union (TSEUR and Immunoprion), the Swiss National Foundation, the National Center for Competence in Research on neural plasticity and repair, and the Ernst Jung Foundation. M.H. is supported by the Bonizzi–Theler Foundation and the Max Cloëtta Foundation. M.H. and M.P. are supported by the Foundation for Research at the Medical Faculty, University of Zürich, Switzerland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Glossary
- Transmissible spongiform encephalopathy
-
(TSE). A synonym for prion disease and a general term that refers to all diseases that are associated with the presence of prions in vacuolated central nervous system tissue. Prions from TSE-affected brains can be transmitted from one affected host to another host.
- Prion
-
An infectious agent with unconventional properties that causes TSE; it is an acronym for 'proteinaceous infectious particle'.
- vCJD
-
(Variant CJD). A type of CJD that is thought to result from the ingestion of beef products that are contaminated with bovine spongiform encephalopathy. The majority of vCJD cases occur in young individuals.
- Iatrogenic CJD
-
CJD transmission from human to human through medical mishaps (for example, transfusion of prion-infected blood).
- Familial CJD
-
Genetic prion diseases that are associated with mutations in one or more loci within the PRNP gene sequence.
- Sporadic CJD
-
The most common CJD, which occurs worldwide at a rate of one case per million people. It mostly affects older adults and the cause is unknown.
- 'Protein-only' hypothesis
-
This hypothesis proposes that the prion is devoid of informational nucleic acid and that the essential pathogenic component is a protein or glycoprotein.
- Scrapie prion protein
-
(PrPSc). An abnormal form of the mature PRNP gene product that is found in tissues of patients with TSE, defined as being partially resistant to proteinase-K digestion under standardized conditions. It is believed to differ from PrPC conformationally and is considered to be the main transmissible agent or prion.
- Cellular prion protein
-
(PrPC). The normally occurring form of the mature PRNP gene product, the presence of which is necessary, but not sufficient, for replication of the prion.
- Prion strain
-
A TSE isolate (or source of infection) that, upon inoculation into genetically identical hosts, causes prion disease with consistent characteristics. The agent-specified information in prion strains is thought to be contained in the distinct conformations of various PrPSc isotypes.
- Horizontal prion transmission
-
The spread of disease between individuals in a certain population or flock of animals. The cause of horizontal prion transmission in some cases (such as scrapie in sheep and chronic wasting disease in elk and deer) remains enigmatic.
- Bovine spongiform encephalopathy
-
(BSE). A TSE that primarily affects cattle, which is believed to be caused by animal feed that was contaminated with the prion agent of either scrapie or BSE. First identified in 1986 in the UK, it became an epidemic that affected hundreds of thousands of cattle in Europe.
- Chronic wasting disease
-
(CWD). A TSE of unknown origin that can be contracted by mule deer, white-tailed deer, Rocky Mountain elk and moose. CWD was identified in the early 1980s in the United States with a horizontal transmission of up to 20%.
- Scrapie
-
A TSE that affects sheep and goats. It has been known since at least the eighteenth century, hundreds of years before prions were first defined. Scrapie was shown to be transmissible 60 years ago.
- Lymphoreticular system
-
Part of the immune system. It is divided into primary (bone marrow and thymus) and secondary lymphoid tissues (spleen and lymph nodes).
- Ancillary genome
-
A putative (secondary) genome within the prion that might carry the information necessary for prion replication and disease phenotype. So far, all evidence points against the presence of an ancillary genome within the prion.
- Vacuolation
-
One of the main neuropathological hallmarks of prion diseases, which results from extensive neuronal loss leading to the occurrence of membrane-lined, optically empty intraneuronal organelles (termed vacuoles) within the brain.
- CJD types
-
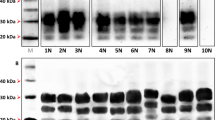
Distinct isoforms of PrPSc that are associated with different CJD phenotypes. CJD types are biochemically distinguished by the different fragment sizes seen on western blots following treatment with proteinase K, as well as the ratio of PrPSc glycoforms and deposition patterns. In this article, we use the CJD classification that was proposed by Gambetti and colleagues.
- Amyloid-β
-
(Aβ). A hydrophobic peptide of 40–42 amino acids and the main component of amyloid plaques in the brains of patients with Alzheimer's disease. Aβ is a product of pathological cleavage of amyloid precursor protein (APP), a transmembrane protein that naturally occurs in the brain and other tissues of mammals.
- Neurotropic
-
Prion strains that mainly attack the CNS. The prion infectivity of individuals who are infected with neurotropic prion strains is primarily contained within the CNS.
- Lymphotropic
-
Prion strains that replicate in the lymphoreticular system before neuroinvasion. The prion infectivity of individuals who are infected with lymphotropic prion strains is found in peripheral lymphoid tissues and the CNS.
- Amyloid deposit
-
A pathologic protein aggregate that occurs in the brains and other tissues of individuals suffering from amyloid or 'protein-misfolding' diseases. The main constituent of amyloid deposits is characteristic for each disease; for example, PrPSc in prion diseases and amyloid-β in Alzheimer's disease.
- Shmerling's disease
-
A neurodegenerative syndrome that occurs in transgenic mice expressing N-terminally truncated PrPC, presenting with ataxia and cerebellar lesions. It can be reversed by the expression of a single allele of full-length PrPC.
- Neuropil
-
The network made out of neuronal processes (axonal, dendritic and glial) within the grey matter of the CNS.
Rights and permissions
About this article
Cite this article
Aguzzi, A., Heikenwalder, M. & Polymenidou, M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8, 552–561 (2007). https://doi.org/10.1038/nrm2204
Issue Date:
DOI: https://doi.org/10.1038/nrm2204
This article is cited by
-
Sporadic Creutzfeldt–Jakob disease infected human cerebral organoids retain the original human brain subtype features following transmission to humanized transgenic mice
Acta Neuropathologica Communications (2023)
-
PMCA screening of retropharyngeal lymph nodes in white-tailed deer and comparisons with ELISA and IHC
Scientific Reports (2023)
-
Vaccines for prion diseases: a realistic goal?
Cell and Tissue Research (2023)
-
PMCA for ultrasensitive detection of prions and to study disease biology
Cell and Tissue Research (2023)
-
Cell biology of prion strains in vivo and in vitro
Cell and Tissue Research (2023)
