
Abstract
In mood disorder, early stressors including parental separation are vulnerability factors, and hippocampal involvement is prominent. In common marmoset monkeys, daily parental deprivation during infancy produces a prodepressive state of increased basal activity and reactivity in stress systems and mild anhedonia that persists at least to adolescence. Here we examined the expression of eight genes, each implicated in neural plasticity and in the pathophysiology of mood disorder, in the hippocampus of these same adolescent marmosets, relative to their normally reared sibling controls. We also measured hippocampal volume. Early deprivation led to decreases in hippocampal growth-associated protein-43 (GAP-43) mRNA, serotonin 1A receptor (5-HT1AR) mRNA and binding ([3H]WAY100635), and to increased vesicular GABA transporter mRNA. Brain-derived neurotrophic factor (BDNF), synaptophysin, vesicular glutamate transporter 1 (VGluT1), microtubule-associated protein-2, and spinophilin transcripts were unchanged. There were some correlations with in vivo biochemical and behavioral indices, including VGluT1 mRNA with reward-seeking behavior, and serotonin 1A receptor mRNA with CSF cortisol. Early deprivation did not affect hippocampal volume. We conclude that early deprivation in a nonhuman primate, in the absence of subsequent stressors, has a long-term effect on the hippocampal expression of genes implicated in synaptic function and plasticity. The reductions in GAP-43 and serotonin 1A receptor expressions are comparable with findings in mood disorder, supporting the possibility that the latter reflect an early developmental contribution to disease vulnerability. Equally, the negative results suggest that other features of mood disorder, such as decreased hippocampal volume and BDNF expression, are related to different aspects of the pathophysiological process.
Similar content being viewed by others


INTRODUCTION
Stressful experiences in early life, such as parental separation or abuse, are significant risk factors for mood disorder (Harris et al, 1986; Heim and Nemeroff, 2001), likely acting in concert with genetic predisposition (Kendler et al, 1993; Jokela et al, 2007). Experimental studies, mostly in rodents, have identified a range of putative pathogenic mechanisms that contribute to the relationship between developmental stressors and later depression-like phenotypes (DeBellis et al, 1999; Sanchez et al, 2001; Pryce et al, 2005a; Ansorge et al, 2007). These include alterations in the hypothalamo-pituitary-adrenal (HPA) axis (Sapolsky, 2000; Gunnar and Quevedo, 2007), the serotonin (5-HT) system (Gartside et al, 2003; Ichise et al, 2006), neurotrophins (Duman et al, 1997), and synaptic plasticity (Manji et al, 2001; Nestler et al, 2002; Pittenger and Duman, 2008). The hippocampus is prominently involved in each of these domains (McEwen, 1999; Sapolsky, 2000; Duman and Monteggia, 2006). Supporting the view that similar processes may be relevant clinically, there is an accumulating literature showing a range of broadly comparable changes in the hippocampus of people with mood disorder, either unipolar or bipolar (for review, see Harrison, 2002; Sala et al, 2004; Czeh and Lucassen, 2007; Frey et al, 2007). These findings include smaller hippocampal volume (Campbell et al, 2004), fewer serotonin 1A receptors (5-HT1AR) (Drevets et al, 1999; Sargent et al, 2000; Lopez-Figueroa et al, 2004; Hirvonen et al, 2008), reductions in synaptic markers (Eastwood and Harrison, 2000; Rosoklija et al, 2000; Webster et al, 2001; Vawter et al, 2002; Tian et al, 2007), and decreased expression of mineralocorticoid and glucocorticoid receptors (Lopez et al, 1998; Webster et al, 2002).
Despite these convergent findings, the molecular links between early life stress and subsequent mood disorder remain poorly understood. One reason has been a paucity of experimental data in the nonhuman primate, requiring extrapolations from the rodent that are compromised by species differences in parenting, endocrine systems, neuroanatomy, and neurodevelopment. Recently, however, a comprehensively assessed primate developmental model of depression vulnerability has been described. In the common marmoset (Callithrix jacchus), a small New World monkey, early deprivation (ED) in the form of daily short periods of isolation from the family group during the first month of life causes endocrine stress responses, and leads to biochemical, cardiovascular, and behavioral effects during juvenility and adolescence, indicative of increased basal activity and reactivity in stress systems, and mild anhedonia (Dettling et al, 2002a, 2002b, 2007; Pryce et al, 2004a, 2004b). These findings as a whole indicate that the ED protocol leads to a ‘prodepressive’ state that is persistent, extending at least to month 12 of life, which is adolescence in this species.
Here we report an initial examination of the long-term neurobiological effects of the ED manipulation, in the same subjects in which the prodepressive in vivo effects were demonstrated. We focused on the hippocampus because of its involvement in stress responses and in the pathophysiology of mood disorder. We measured the expression of eight genes selected in light of reported findings in subjects with mood disorder, and because they serve as markers of processes implicated in the pathophysiological links between stress and depression: synaptophysin, growth-associated protein-43 (GAP-43), brain-derived neurotrophic factor (BDNF), microtubule-associated protein-2 (MAP-2), spinophilin, vesicular glutamate transporter 1 (VGluT1), vesicular GABA transporter (VGAT, also called VIAAT), and the serotonin 1A receptor, as well as estimating hippocampal volume. We also inspected for correlations between these neurobiological parameters measured in adolescence, and the in vivo biochemical and behavioral variables that were affected by ED.
MATERIALS AND METHODS
The animals were bred, and all in vivo studies conducted, at the Laboratory for Behavioural Neurobiology of the Swiss Federal Institute of Technology, Zurich, under experimental permit in accordance with the Swiss Animal Protection Act (1978). The brains were shipped to Oxford for study under license from the Convention for International Trade in Endangered Species of Wild Fauna and Flora, administered by the Swiss Federal Office for Veterinary Affairs and the UK Department for Environment, Food and Rural Affairs.
The Early Deprivation Intervention
The marmoset is characterized by monogamous breeding, dizygotic twins, and high levels of caregiving by both parents. In this study, each of nine sets of parents contributed, in random order, one pair of ED twins and one pair of control twins successively, with an interval of 4–6 weeks between termination of the study with the first pair and the birth of the second pair; one set of parents contributed one pair of ED twins only. The total sample was therefore 10 pairs of ED twins and 9 pairs of control twins (Table 1). The ED intervention and its in vivo sequelae have been described in detail previously (Dettling et al, 2002a, 2002b, 2007; Pryce et al, 2004a, 2004b). Briefly, ED infants were separated from their parents for 30–120 min each day, using variable durations as well as times of day, on postnatal days 2–28. The ED was carried out consecutively within each twin pair, such that one infant remained with the parents at all times. Controls were briefly handled on the back of the carrying parent on days 2–28. After day 28, subjects remained in the home cage with the family group, and there were no further interventions that differed between ED and control subjects. Additional manipulations were: at age 15 weeks, in three families, subjects were surgically operated to implant a radiotransmitter for the measurement of blood pressure; following a 2-week recovery period, endocrine and behavioral measures of these subjects were within the range of other subjects. At age 18–20 weeks, in all families, subjects were studied in six 60-min tests of isolation from the family in a novel physical environment. Otherwise, all behavioral testing, and the collection of physiological samples, were carried out in the home cage.
At age 48 weeks (range: 43–51 weeks), animals were euthanized. For each group, one subject was removed from the home cage to a procedures room, sedated (Saffan, 10 mg/kg, i.m.), and physiological samples were collected. The animal was then deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.), and the brain either removed and frozen immediately (for the molecular studies), or the animal was transcardially perfused (for hippocampal volumetry; see below). At 1–4 days later, the procedure was repeated with the second twin. Two subjects (two control males) were euthanized at 16 and 38 weeks due to injury and excluded from the present study. The frozen brains comprised 11 ED (7 male and 4 female), and 9 controls (5 male and 4 female); the fixed brains comprised 7 ED (4 male and 3 female), and 6 controls (3 male and 3 female, with 3 brains not studied due to poor perfusion). All brains were coded, and all experiments conducted and analyzed blind to group status.
Brain Processing and Sectioning
The brains to be frozen were rinsed with cold saline, immersed in isopentane at −50°C and stored at −80°C. After warming to −20°C, each brain was serially sectioned on a cryostat in the coronal plane at 10 μm, with two adjacent sections mounted onto each slide, and stored at −80°C. Every 20th slide was taken for Nissl staining, and used to identify the anterior and posterior limits of the hippocampus. Within these limits, for each ISH experiment, the hippocampus was sampled at ∼2 mm intervals, giving three hippocampal levels (anterior, middle, and posterior), with two sections at each level, per animal.
For brains to be fixed, animals were transcardially perfused with saline for 2 min followed by 4% paraformaldehyde at 4°C for 12 min. The head was removed and fixed in 4% paraformaldehyde at 4°C for 24 h. The brain was then transferred to 10% sucrose solution for 1 × 24 h at 4°C, and to 30% sucrose solution for 1 × 24 h, and then stored at −80°C. These brains were cut at 50 μm on a Leica sliding microtome using solid carbon dioxide, and collected in vials containing cryoprotectant (300 ml glycine, 300 ml ethylene glycol, 400 ml distilled water, 5.4 g disodium hydrogen orthophosphate, and 1.57 g sodium dihydrogen orthophosphate per liter). Every fifth section was mounted on 2% gelatine-treated slides, stained with cresyl violet and coverslipped.
In Situ Hybridization with Oligonucleotides
With the exception of BDNF (see below), in situ hybridization was performed with olignonucleotide probes. As marmoset cDNA sequences were not available, the proposed target region of each transcript was amplified from marmoset cDNA using RT–PCR with primers designed to the human cDNA sequence. RT–PCR products were then sequenced, and oligonucleotide probes designed that were 100% complementary to the marmoset cDNA sequence (primers and sequences available on request).
For in situ hybridization, the frozen sections were thawed, fixed, acetylated, and delipidated (Law et al, 2003). Oligonucleotides were 3′ end labeled with [35S]dATP (1250 Ci/mmol; Perkin Elmer, UK) in a 10 : 1 molar ratio using terminal deoxynucleotidyl transferase (Promega, UK). The experimental conditions and film exposure times for each probe were optimized in pilot studies; the definitive experiment for each transcript was performed in a single experimental run. Sections were incubated overnight at 40–42°C with hybridization buffer containing 1.0 × 106 c.p.m. of labeled probe, as described (Law et al, 2003). Postincubation washes were carried out in 1 × SSC at 55°C for 3 × 20 min and 1 h at room temperature. Experimental controls comprised: hybridization with sense-strand probes, hybridization in the presence of 50-fold excess unlabeled probe, and ribonuclease (RNase A 200 μg/ml at 37°C for 20 min) pretreatment. After air drying, slides were apposed to autoradiographic film (Kodak, Rochester, NY, USA) along with 14C microscales (Amersham Pharmacia Biotech, Sweden) for 3 weeks (GAP-43 and serotonin 1A receptor), 2 weeks (synaptophysin, spinophilin and VGAT), or 1 week (VGluT1 and MAP-2).
Riboprobe In situ Hybridization For BDNF
BDNF mRNA was detected using a riboprobe, for which a 1.6 kb segment of the human BDNF cDNA (accession no. M61176) was cloned into Bluescript vector (Stratagene) to generate a cDNA template. The specificity of this clone was previously confirmed by northern blotting (Weickert et al, 2003). Sense and antisense riboprobes for BDNF were generated from linearized plasmids using a T7 or T3 polymerase (according to insert orientation), an in vitro transcription kit as recommended by the manufacturer (Promega) and 35S-UTP (Amersham Pharmacia Biotech), labeled to a specific activity of ∼1.6 × 109 c.p.m./μg and purified by ethanol precipitation. The sequence recognized by this clone is contained within the common BDNF exon (human chromosome 11: 27635676–27637249) and spans the entire protein coding domain. For BDNF in situ hybridization, two hippocampal sections per animal were thawed, fixed in 4% formalin in 0.1 M PBS, acetylated, and dehydrated as described (Whitfield et al, 1990). Sections were hybridized with 200 μl of hybridization buffer containing 5 ng/ml of radiolabeled probe at 55°C overnight in a humidified chamber. RNase digestions and stringent post-hybridization washes were performed as described (Whitfield et al, 1990). After air drying, slides were apposed to film (Kodak Biomax) for 2 weeks along with 14C microscales.
Receptor Autoradiography with [3H]WAY100635
Autoradiographic analysis of serotonin 1A receptor-binding site densities was carried out using [3H]WAY100635 as described (Burnet et al, 1997). Briefly, sections were thawed and preincubated at room temperature in 50 mM Tris-HCl buffer (pH7.4) for 30 min, then incubated in 50 mM Tris-HCl containing 3 nM [3H]WAY100635 for 2 h at room temperature. Nonspecific binding was determined by incubation of adjacent sections with 10 μM 5-HT. Slides were washed with the same buffer for 2 × 4 min at 4°C. Sections were air dried and apposed to Biomax film for 3 weeks.
Image Analysis and Statistical Analysis
Four hippocampal subfields (dentate gyrus, CA3, CA1, and subiculum) were identified, by reference to a marmoset brain atlas (Stephan et al, 1980). Pilot studies did not show consistent rostro–caudal differences in hippocampal expression of any of the genes; hence, autoradiographic signals were measured in each subfield, across all hippocampal sections, and the mean value for each subfield was used for the statistical analysis. All analyses were blind to ED or control status.
Data were examined for normality using the Kolmogorov–Smirnov one-sample test. For each gene, comparisons between ED and control animals were made using a two-stage approach. First, by repeated-measures ANOVA (RM-ANOVA) with subfield as within-subjects factor, and group (ED or control) and sex as between-subjects factors. Sex was included because of the prior evidence for sexual dimorphism in hippocampal gene expression (eg Lustig et al, 1993; Zhang et al, 1999), in animal models of depression and stress, and in the clinical vulnerability to mood disorder (see ‘Discussion’). Where the RM-ANOVA revealed a main effect of ED, or an ED-by-subfield interaction, group differences were examined in each subfield separately using one-way ANOVA. Analyses were carried out using SPSS for Windows (v.15.0).
Estimation of Hippocampal Volume
Hippocampal volume was estimated in cresyl violet-stained sections from the perfusion-fixed brains using point counting and Cavalieri's theorem (Walker et al, 2002). A grid, 25 × 25 mm, was thrown over all sections containing the hippocampus from each animal (mean 24 sections, range: 15–32), and the cross-sectional area of the hippocampus, excluding the subiculum, estimated. The sections were spaced 200 μm apart. No correction was made for tissue shrinkage during processing. Left and right hippocampi were measured separately. Data were analyzed using RM-ANOVA with hemisphere as the within-subjects factor and group and sex as between-subjects factors.
Correlations with In Vivo Measures
We explored whether the parameters measured here were related to three in vivo biochemical and three behavioral indices that were affected by the ED intervention (see Table 1) using Spearman's correlations. As a partial control against multiple testing, we set α=0.02 and followed up each significant correlation by examining whether it persisted after partialling for the effect of ED; we also inspected whether a similar correlation was present in control and ED groups considered separately.
RESULTS
Each transcript was successfully detected (Figures 1 and 2). Specificity of signal was confirmed using the sense probes, cold displacement, and RNase, which produced negligible signals (not shown). The regional distribution of each mRNA, and for [3H]WAY100635 binding, was consistent with findings in the hippocampus of other species (see ‘Discussion’).

Distribution of five presynaptic protein transcripts in the marmoset hippocampus. (a) Growth-associated protein-43 (GAP-43) mRNA. (b) Synaptophysin mRNA. (c) Vesicular glutamate transporter 1 (VGluT1) mRNA. (d) Vesicular GABA transporter (VGAT) mRNA. (e) Brain-derived neurotrophic factor (BDNF) mRNA. Abbreviations: DG, dentate gyrus; SUB, subiculum.

Distribution of four postsynaptically expressed genes in the marmoset hippocampus. (a) Microtubule-associated protein-2 (MAP-2) mRNA. (b) Spinophilin mRNA. (c) Serotonin 1A receptor mRNA. (d) [3H]WAY100635 binding.
Effect of Early Deprivation on Expression of Presynaptic Genes
For GAP-43 mRNA (Figure 3a), there was an ED-by-subfield interaction (F(3,48)=2.97, p=0.041), with a trend overall effect of ED (F(1,16)=3.40, p=0.084) but no effect of sex or ED-by-sex interaction. GAP-43 mRNA was decreased after ED in the dentate gyrus (F(1,18)=4.52, p=0.048) and CA3 (F(1,18)=5.42, p=0.031), but not in CA1 or subiculum.

Expression of presynaptic genes in the marmoset hippocampus, in animals subjected to early deprivation (ED, diamonds and controls, squares). (a) Growth-associated protein-43 (GAP-43) mRNA. (b) Synaptophysin mRNA. (c) Vesicular GABA transporter (VGAT) mRNA. (d) Vesicular glutamate transporter 1 (VGluT1) mRNA. (e) Brain-derived neurotrophic factor (BDNF) mRNA. Abbreviations: DG, dentate gyrus; Sub, subiculum. *p<0.05, **p<0.01.
Synaptophysin mRNA (Figure 3b) was unaffected by ED and showed no interactions with subfield or sex. There was a trend effect of sex (p=0.067), which reflected higher levels of synaptophysin mRNA in females than males in CA3 (p=0.039) and CA1 (p=0.045).
VGAT mRNA (Figure 3d) showed an ED-by-subfield interaction (F(3,48)=2.93, p=0.043) and a trend overall effect of ED (F(1,16)=4.30, p=0.055), but no effect of sex or ED-by-sex interaction. VGAT mRNA was increased after ED in CA3 (F(1,18)=9.28, p=0.007) and with a trend in dentate gyrus (F(1,18)=4.16, p=0.056), but not in CA1 or subiculum.
VGluT1 mRNA (Figure 3c) and BDNF mRNA (Figure 3e) did not differ after ED or between the sexes, and there were no subfield interactions.
Effect of Early Deprivation on Expression of Postsynaptic Genes
MAP-2 mRNA (Figure 4a) and spinophilin mRNA (Figure 4b) showed no effects of ED, sex, nor an ED-by-subfield interaction.

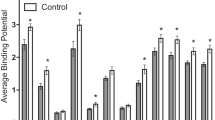
Expression of postsynaptic genes in the marmoset hippocampus, in animals subjected to early deprivation (ED, diamonds and controls, squares). (a) Serotonin 1A receptor mRNA. In addition to the decrease in CA1 in the ED group, there are ED-by-sex interactions in DG and CA3 (see text). (b) [3H]WAY100635 binding. In addition to the decrease in CA1 in the ED group, there is an ED-by-sex interaction in CA3 (see text). (c) Microtubule-associated protein-2 (MAP-2) mRNA. (d) Spinophilin mRNA. One animal in the ED group had spinophilin mRNA values >2 SD greater than all others; there remains no difference between ED and control groups when it is omitted. Abbreviations: DG, dentate gyrus; Sub, subiculum. *p<0.05.
Effect of Early Deprivation on Expression of Serotonin 1A Receptors
For serotonin 1A receptor mRNA (Figure 4c) there was an ED-by-sex interaction (F(1,16)=4.98, p=0.04) and an ED-by-sex-by-subfield interaction (F(2,32)=3.76, p=0.034). (The subiculum was excluded from the serotonin 1A receptor RM-ANOVA due to missing data). Subsequent analysis showed that, in CA1, serotonin 1A receptor mRNA was decreased following ED (F(1,16)=6.41, p=0.022) with no sex interaction. There was an ED-by-sex interaction in the dentate gyrus (F(1,16)=5.10, p=0.038) and in CA3 (F(1,6)=6.74, p=0.019). In both subfields, serotonin 1A receptor mRNA tended to be decreased after ED in females but increased in males, but the post hoc tests in each sex were not significant (p=0.07–0.16). For example, in CA3, control vs ED: females, 66±4 vs 52±4 nCi/g (t=1.63, p=0.15); males, 54±5 vs 66±3 (t=2.03, p=0.07).
The density of [3H]WAY100635-binding sites showed an ED-by-subfield interaction (F(3,48)=3.01, p=0.039) but no interaction with sex. [3H]WAY100635 binding was decreased by ED in CA1 (F(1,16)=4.70, p=0.046) with no change in dentate gyrus or subiculum (Figure 4d). In CA3, there was an ED-by-sex interaction (F(1,16)=5.62, p=0.031); post hoc tests showed increased CA3 [3H]WAY100635 binding in males after ED (controls vs ED: 16.2±1.0 vs 19.4±1.0 nCi/g, t=2.30, p=0.043) with no alteration in females (controls vs ED: 16.4±2.0 vs 18.9±0.6 nCi/g, t=1.21, p=0.27).
Effect of Early Deprivation on Hippocampal Volume
For hippocampal volume, there was an effect of sex (F(1,19)=5.59, p=0.042), with the hippocampus being larger in females (54.3±2.5 mm3) than males (46.2±2.3 mm3). However, there was no effect of ED, no ED-by-sex interaction, and no left–right difference (Table 2).
Correlations with In Vivo Measures
Several of the exploratory correlations between the in vivo measures and adolescent gene expression met our criteria for significance. (a) Social play in infancy, which is decreased by ED (Dettling et al, 2002a), correlated inversely with [3H]WAY100635 binding in CA3 (Figure 5a; r=−0.606, n=20, p=0.005; partialling for ED: r=−0.566, d.f.=17, p=0.011), and with a similar trend in ED and control groups (respectively, r=−0.790, n=11, p=0.004; r=−0.594, n=9, p=0.09). (b) In adolescence, ED marmosets exhibited reduced motivation for palatable reward in terms of fewer operant responses on a progressive ratio reinforcement schedule (Pryce et al, 2004a), and this measure of mild anhedonia correlated with VGluT1 mRNA in CA1 (Figure 5b; r=0.707, n=15, p=0.003; partialling for ED: r=0.655, d.f.=12, p=0.011); the correlation was present in ED (r=0.719, n=8, p=0.045) and control (r=0.893, n=7, p=0.007) groups. (c) Urinary noradrenaline is raised by ED, from late infancy to adolescence (Pryce et al, 2004a), and average levels correlated negatively with synaptophysin mRNA (Figure 5c; r=−0.733, n=11, p=0.01), with a similar finding in ED (r=−0.899, n=6, p=0.015) and control (r=−0.700, n=5, p=0.18) groups. (d) Finally, terminal (ie adolescent) CSF cortisol level correlated with serotonin 1A receptor mRNA, especially in the dentate gyrus (Figure 5d; r=0.530, n=20, p=0.02). The correlation survived partialling for the effect of ED (r=0.503; d.f.=16; p=0.033), sex (r=0.519; d.f.=16; p=0.027), or both (r=0.496; d.f.=15; p=0.043). The correlation was present in the ED group (r=0.818, n=11, p=0.002) but not in the controls (r=0.095, n=8, p=0.82).

Correlations between gene expression and in vivo measures. (a) [3H]WAY100635 binding in CA3 correlates inversely with social play during infancy (r=−0.606, p=0.005, n=20). (b) Vesicular glutamate transporter 1 (VGluT1) mRNA in CA1 correlates with the number of progressive ratio reinforcements, a measure of motivation for reward (r=0.707, p=0.003, n=15). (c) Dentate gyrus synaptophysin mRNA correlates inversely with urinary noradrenaline, measured in males only (r=−0.733, p=0.01, n=11). (d) Dentate gyrus serotonin 1A receptor (5-HT1AR) mRNA correlates with CSF cortisol (r=0.530, p=0.02, n=20).
Hippocampal volume did not correlate with any of the in vivo measures, and neither impulsivity nor urinary dopamine correlated with expression of any of the genes.
DISCUSSION
As noted in the Introduction, early life stressors in rodents, such as maternal separation, produce long-term biochemical, endocrine, and behavioral alterations. These complement, and may help explain, the epidemiological findings that early life adversity confers an increased risk of mood and emotional disorders later in life in humans. The hippocampus is centrally implicated in the response to early life adversity, and in mood disorder. Here, we studied a recently validated nonhuman primate model of depression vulnerability to explore the hypothesis that ED would produce long-term alterations in the expression of genes implicated in hippocampal synaptic plasticity and functioning, and in the pathophysiology of mood disorder. Our hypothesis was supported by some findings (eg decreased GAP-43 and serotonin 1A receptor expression) but not others (eg unchanged BDNF mRNA and hippocampal volume).
Expression Profiles of the Genes in the Marmoset Hippocampus
Each transcript showed a regional distribution within the marmoset hippocampus that was as anticipated from in situ hybridization studies in other primates and in rodents. The profile for [3H]WAY100635 with enhancement in CA1 was also as expected (Burnet et al, 1997). Here we note two points of interest.
In the rodent, GAP-43 mRNA is expressed by neurons in the hippocampus proper but is barely detectable in the dentate gyrus (McNamara et al, 1996). In contrast, in the macaque (Higo et al, 1998) and human (Eastwood and Harrison, 1998), GAP-43 mRNA is strongly expressed by granule cells of the dentate gyrus; indeed, it is more abundant therein than in other hippocampal subfields. The New World marmoset appears in this respect to lie between the rodent and these higher Old World primate species, with signal for dentate gyrus GAP-43 mRNA being robust, but less than that seen over CA3.
The VGAT mRNA dentate gyrus signal was shown in emulsion-dipped sections to arise from expression by hilar neurons adjacent to the granule cell layer (data not shown); similarly, the punctate VGAT mRNA signal in the other subfields arose from scattered medium-sized cells rather than from the pyramidal neurons. This pattern is the same as that observed in the rat hippocampus (McIntire et al, 1997) and is consistent with VGAT mRNA being expressed by GABAergic neurons, in line with its role as the vesicular GABA transporter.
Effect of Early Deprivation on Expression of Synaptic Protein Genes
Hippocampal plasticity in rodents is affected by maternal care, rearing conditions and by other developmental stressors (Liu et al, 2000; Andersen and Teicher, 2004; Bogoch et al, 2007). One manifestation is a decreased expression of genes that serve as markers of presynaptic terminal density, activity, and/or plasticity, such as synaptophysin and GAP-43 (Andersen and Teicher, 2004; Bredy et al, 2004; Reines et al, 2008). The same approach has been taken to assess whether similar processes occur in the hippocampus of subjects with mood disorder, and the data show reductions in expression of several presynaptic proteins (Eastwood and Harrison, 2000; Fatemi et al, 2001; Webster et al, 2001; Vawter et al, 2002; Tian et al, 2007). Here, we found that ED decreases hippocampal expression of GAP-43, suggestive of a long-term effect on hippocampal synaptic plasticity, at least as indicated by this marker (Benowitz and Routtenberg, 1997). In contrast, synaptophysin expression did not change following ED. Synaptophysin is usually considered a marker of presynaptic terminal density rather than plasticity (Harrison and Eastwood, 2001), and as such our data suggest that overall hippocampal synaptic density is not markedly altered by ED. However, dentate gyrus synaptophysin expression was inversely correlated with level of urinary noradrenaline, which is increased in ED marmosets (Pryce et al, 2004a) as well as in depressed children with a history of early life adversity (DeBellis et al, 1999), and is indicative of sympathoadrenal hyperactivity. This suggests that presynaptic terminal density may be important for the hippocampus in regulating autonomic functions (Fuchs et al, 1995).
VGluT1 and VGAT are vesicular transporters for glutamate and GABA, respectively. Effects on both transmitters have been reported after other developmental interventions and stressors in rats (eg Hsu et al, 2003; Pickering et al, 2006) and hippocampal VGluT1 expression is modulated by antidepressants (Tordera et al, 2005). In mood disorder, there are emerging data to support both glutamatergic (Choudary et al, 2005; Yildiz-Yesiloglu and Ankerst, 2006; Hashimoto et al, 2007) and GABAergic (Brambilla et al, 2003; Taylor et al, 2003) dysfunction. Following ED, we found that VGAT mRNA was increased whereas VGluT1 mRNA was unchanged. Importantly, there is a quantitative relationship between the expression of these transporters and the release of their transmitter (De Gois et al, 2005; Wilson et al, 2005); moreover, vesicular loading by VGAT is directly coupled to GABA synthesis (Jin et al, 2003). Hence, the elevation of VGAT expression following ED suggests an enhancement of hippocampal GABA signaling, a possibility that requires direct investigation. GABA signaling is altered in rodents following developmental stressors (Hsu et al, 2003; Caldji et al, 2000) and in subjects with mood disorder (Brambilla et al, 2003; Taylor et al, 2003; Frey et al, 2007), including altered hippocampal expression of GABAergic genes (Sequeira et al, 2007). However, these prior findings tend to implicate impaired GABA functioning. As such, the increased VGAT expression observed here may be an adaptive response to the effects of ED on other determinants of GABA signaling. If so, then extrapolating to humans, perhaps enhanced GABA functioning (including increased VGAT) is protective, and that depression occurs when this compensatory process fails. To test this hypothesis, it would be of interest to assess GABA function in healthy subjects at risk of mood disorder by virtue of their exposure to early life stressors, and to follow them longitudinally and study the effect of a first depressive episode. Unchanged expression of VGluT1 suggests that major alterations in glutamate release do not occur after ED but, in the context of enhanced VGAT expression, implies that the balance between inhibitory and excitatory hippocampal transmission is affected. Also, the correlation with responses on the test of motivation for palatable reward that revealed mild anhedonia in ED adolescents provides an indication that hippocampal VGluT1 expression may be functionally relevant (ie with regard to anhedonia) even in the absence of an overall effect of ED. In rodent models of anhedonia induced by chronic stress in adulthood, the hippocampus is certainly a major region of altered activity and gene expression (Holderbach et al, 2007; Bergstrom et al, 2007). In mood disorder, hippocampal VGluT1 mRNA is unaltered (McCullumsmith and Meador-Woodruff, 2003) and VGAT expression has not been reported.
In addition to studying these presynaptic genes, we utilized molecular markers of dendrites and dendritic spines, namely MAP-2 and spinophilin respectively, to assess the postsynaptic component of synaptic plasticity. In mood disorder, there are preliminary data showing a reduction in spinophilin expression (Law et al, 2004) and for morphological alterations in hippocampal dendrites and spine densities (Rosoklija et al, 2000). However, no differences in expression of either MAP-2 or spinophilin were seen here, and as such we found no evidence to support an alteration in postsynaptic plasticity after ED. Undoubtedly many other genes are involved in dendritic plasticity, and there is much more to the process than simply changes in gene expression. Nevertheless, the discrepancy following ED between the changes seen presynaptically, and the lack of change postsynaptically, is notable; it was also seen in a microarray study of the hippocampal response to prenatal stress in rodents (Bogoch et al, 2007).
Effect of Early Deprivation on Serotonin 1A Receptors
Serotonin is involved in neurodevelopment (Whitaker-Azmitia, 2001), in stress responses (Chaouloff et al, 1999), and in the pathophysiology of mood disorder (Deakin, 1998; Mann, 1999), with the serotonin 1A receptor being important in all these situations (Cowen et al, 1994; Sibille and Hen, 2001; Strobel et al, 2003). Three neuroimaging studies of mood disorder show decreased hippocampal serotonin 1A receptor binding (Drevets et al, 1999; Sargent et al, 2000; Hirvonen et al, 2008), and hippocampal serotonin 1A receptor mRNA is also decreased (Lopez-Figueroa et al, 2004). Reduced hippocampal serotonin 1A receptor binding has also been seen in long-tailed macaques with behavioral depression (Shively et al, 2006), and serotonin 1A receptor expression and/or function are decreased by some experimental stressors in rodents (Lopez et al, 1998; Van Riel et al, 2004; Griffin et al, 2005). Moreover, a polymorphism in the serotonin 1A receptor gene has been associated with depression-related personality traits and with lower postsynaptic serotonin 1A receptor expression (Strobel et al, 2003; Czesak et al, 2006).
Although hippocampal serotonin 1A receptor reductions have not always been replicated in mood disorder (Stockmeier, 2003; Parsey et al, 2006), or following maternal separation in rodents (Neumaier et al, 2002), the evidence as a whole led us to predict that ED would result in lowered hippocampal serotonin 1A receptor expression. This is what was observed, in CA1, for both serotonin 1A receptor mRNA and receptor binding. Our results suggest that a diminished serotonin 1A receptor availability, particularly in CA1, may be an ‘at risk’ marker for mood disorder, consistent with the fact that hippocampal serotonin 1A receptor binding remains decreased after recovery from depression (Sargent et al, 2000; Bhagwagar et al, 2004), and that it may arise as a result of early adverse experiences.
Apart from CA1, interpretation of the serotonin 1A receptor findings is complicated by the interactions between ED and sex. In CA3 and dentate gyrus, only females showed decreases in serotonin 1A receptor mRNA and [3H]WAY100635 binding, whereas males tended to show increases. Although the post hoc statistical tests for males and females separately were not conclusive (reflecting the small sample size), these findings are in line with other results suggestive of sexual dimorphisms, affecting relationships between serotonin 1A receptor function, stress, and mood disorder. For example, neonatal handling affects hippocampal serotonin 1A receptors more in female than male rats (Stamatakis et al, 2006), and females have a greater vulnerability to mood disorder clinically (Kendler et al, 2006) and in animal models of depression (Faraday, 2002; Barna et al, 2003). If our finding extrapolates to humans, it might indicate that women are more prone to serotonin 1A receptor downregulation in response to environmental stressors, and that this may play a role in their greater vulnerability to mood disorder.
If hippocampal serotonin 1A receptor reduction is part of the molecular mechanisms that mediate between early life stress and an enhanced risk of mood disorder, one co-candidate is HPA axis dysfunction. Hippocampal serotonin 1A receptor expression is regulated by corticosteroids, with elevated levels of the latter typically reducing expression of the former (Burnet et al, 1992; Chalmers et al, 1993; Le Corre et al, 1997; Meijer et al, 1997; Neumaier et al, 2000; but see Montgomery et al, 2001), in part by transcriptional regulation (Meijer et al, 2000). However, we found no inverse correlations between cortisol and serotonin 1A receptor mRNA or binding; indeed the only correlation we did observe was positive (Figure 5d). Of note, ED induces marked acute increases in cortisol in infants (Dettling et al, 2002a), but in juvenility and adolescence the elevations are modest and are not seen in morning samples but only in afternoon samples (Pryce et al, 2004b; Dettling et al, 2007). This suggests that any contribution of ED-induced hypercortisolism to altered serotonin 1A receptor levels occurred in infancy; perhaps the acute increases in cortisol in response to ED influenced the long-term set-point of serotonin 1A receptor expression (see Ansorge et al, 2007). Further study is required to establish the interplay between HPA axis alterations and serotonin 1A receptor reductions, and the relationship of both to the effects of ED.
There was an inverse correlation between social play in infancy and CA3 serotonin 1A receptor binding in adolescence, which suggests that the reduced social play observed in ED infants might have been related to increased 5-HT function, as reported for peri-adolescent rats (Homberg et al, 2007).
Effect of Early Deprivation on BDNF Expression
The prominent BDNF hypothesis of mood disorder postulates that a deficiency of BDNF is important in its pathophysiology, and that BDNF normalization is a key part of the therapeutic mechanism of antidepressants (Duman et al, 1997; Hashimoto et al, 2004). The theory has a firm preclinical basis, in that there is much evidence for a critical role of BDNF in neurodevelopment and plasticity (Huang and Reichardt, 2001), and for hippocampal BDNF being reduced after a range of stressors (Duman and Monteggia, 2006), including in adult rats that had been subjected to brief maternal deprivation (Roceri et al, 2002). The roles of BDNF in human hippocampal function are also not in doubt (Egan et al, 2003). In depressed subjects, several studies show reduced serum BDNF levels (see Duman and Monteggia, 2006), and two found decreased hippocampal BDNF expression in suicide victims, many of whom were depressed (Dwivedi et al, 2003; Karege et al, 2005), whereas another reported higher hippocampal BDNF immunoreactivity in depressed subjects on antidepressants at death compared to those who were not (Chen et al, 2001).
Given these findings, we predicted that BDNF expression would be lower following ED, reflecting the intervention itself and its prodepressive consequences. However, we found no changes in BDNF mRNA. Notably, contrary to Roceri et al (2002), rodent studies of repeated maternal separation find hippocampal BDNF expression in adulthood to be unchanged (Roceri et al, 2004) or increased (Greisen et al, 2005), or to differ between splice variants (Nair et al, 2006). These inconsistent findings likely reflect the complex temporal and molecular regulation of BDNF expression (Pruunsild et al, 2007), and other factors such as the timing and duration of the intervention, and the interval between the intervention and measurement (Pryce et al, 2005a; Lippmann et al, 2007). Any of these variables might explain our negative BDNF result. With regard to mood disorder, the present finding raises the possibility that reduced BDNF in that situation is independent of the early stressor component of pathogenesis. For example, it might be part of a genetic predisposition, or not emerge until a significant depressive episode occurs. In any event, there is also a negative study of hippocampal BDNF expression in mood disorder (see Knable et al, 2004), and criticism of the ‘deficient BDNF hypothesis’ itself (Groves, 2007).
Effect of Early Deprivation on Hippocampal Volume
We had anticipated that the ED intervention might lead to hippocampal atrophy, given that human imaging studies have reported smaller hippocampi not only in mood disorder (Campbell et al, 2004) but also in people who had experienced severe early life stress, regardless of their later psychiatric diagnosis (Vythilingam et al, 2002; Geuze et al, 2005). However, not all such studies have been positive (Cohen et al, 2006; Lenze et al, 2008) and we did not find any effect of ED on hippocampal volume. This finding supports the view that hippocampal atrophy in mood disorder is not related to the early developmental component of pathogenesis, but is a correlate or consequence of the disorder (Czeh and Lucassen, 2007). This interpretation is consistent with the fact that reduced hippocampal volume is best established in those with chronic or recurrent depressive episodes (Sheline et al, 1996; Campbell et al, 2004). Although the ED animals exhibited decreased motivation for reward, suggestive of mild anhedonia (Pryce et al, 2004b), they did not exhibit a true depression-like phenotype. In the ED paradigm, stress is confined to a specific period and thereafter subjects remain undisturbed in the family group: it would be of interest to assess whether hippocampal volume would be reduced if ED were combined with a subsequent stressor applied in adolescence. Finally, there may be a genetic predisposition to smaller hippocampal volume in mood disorder (Frodl et al, 2007), which would not be captured here.
CONCLUSIONS
The present study shows that early life stress in a nonhuman primate produces long-term effects on hippocampal gene expression, suggestive of alterations in synaptic functioning and plasticity. The correlations seen between gene expression and the in vivo behavioral and biochemical effects of ED speak to the potential functional significance of the molecular alterations. The congruence between the ED effect and observations in mood disorder, especially the reduction in serotonin 1A receptors, suggests that hippocampal involvement in the disorder begins prior to the first illness episode, and may reflect at least partly the role that early developmental adversity plays in its origins. Conversely, the lack of effect of ED on other indices, such as hippocampal volume and BDNF mRNA, suggests that their involvement in mood disorder arises from different aspects of the pathogenic process, or may be secondary to the effects of illness or its treatment.
Clearly, many further issues await investigation in this marmoset model of the long-term effects of early life stress. First, to measure the encoded proteins; this was not possible here due to tissue limitations. Second, to ascertain the role that HPA axis alterations, notably glucocorticoid and mineralocorticoid receptors, play in the response to ED (Pryce et al, 2005b). Third, to examine other limbic regions such as the medial prefrontal cortex, wherein comparable changes related to stress (Cohen et al, 2006; Ichise et al, 2006; Burton et al, 2007) and mood disorder (Manji et al, 2001; Harrison, 2002; Choudary et al, 2005) also occur. Finally, expression profiling (Sabatini et al, 2007) and study of the cellular changes that may follow ED, such as altered hippocampal neurogenesis (Sahay and Hen, 2007), will help paint a more detailed picture.
Accession codes
References
Andersen SL, Teicher MH (2004). Delayed effects of early stress on hippocampal development. Neuropsychopharmacology 29: 1988–1993.
Ansorge MS, Hen R, Gingrich JA (2007). Neurodevelopmental origins of depressive disorders. Curr Opin Pharmacol 7: 8–17.
Barna I, Bálint E, Baranyi J, Bakos N, Makara GB, Haller J (2003). Gender-specific effect of maternal deprivation on anxiety and corticotropin-releasing hormone mRNA expression in rats. Brain Res Bull 62: 85–91.
Benowitz LI, Routtenberg A (1997). GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci 20: 84–91.
Bergstrom A, Jayatissa MN, Thykjaer T, Wiborg O (2007). Molecular pathways associated with stress resilience and drug resistance in the chronic mild stress rat model of depression: a gene expression study. J Mol Neurosci 33: 201–215.
Bhagwagar Z, Rabiner EA, Sargent PA, Grasby PM, Cowen PJ (2004). Persistent reduction in brain serotonin1A receptor binding in recovered depressed men measured by positron emission tomography with [11C]WAY-100635. Mol Psychiatry 9: 386–392.
Bogoch Y, Biala YN, Linial M, Weinstock M (2007). Anxiety induced by prenatal stress is associated with suppression of hippocampal genes involved in synaptic function. J Neurochem 101: 1018–1030.
Brambilla P, Perez J, Barale F, Schettini G, Soares JC (2003). GABAergic dysfunction in mood disorders. Mol Psychiatry 8: 721–737.
Bredy TW, Zhang TY, Grant RJ, Diorio J, Meaney MJ (2004). Peripubertal environmental enrichment reverses the effects of maternal care on hippocampal development and glutamate receptor subunit expression. Eur J Neurosci 20: 1355–1362.
Burnet PWJ, Eastwood SL, Harrison PJ (1997). [3H]WAY-100635 for 5-HT1A receptor autoradiography in human brain: a comparison with [3H]8-OH-DPAT and demonstration of increased binding in the frontal cortex in schizophrenia. Neurochem Int 30: 565–574.
Burnet PWJ, Mefford IN, Smith CC, Gold PW, Sternberg EM (1992). Hippocampal 8-[3H]hydroxyl-2-(di-n-propylamino) tetralin binding site densities, serotonin receptor (5-HT1A) messenger ribonucleic acid abundance, and serotonin levels parallel the activity of the hypothalamo-pituitary-adrenal axis in rat. J Neurochem 59: 1062–1070.
Burton CL, Chatterjee D, Chakraborty M, Lovic V, Grella SL, Steiner M et al (2007). Prenatal restraint stress and motherless rearing disrupts expression of plasticity markers and stress-induced corticosterone release in adult female Sprague-Dawley rats. Brain Res 1158: 28–38.
Caldji C, Francis D, Sharma S, Plotsky PM, Meaney MJ (2000). The effects of early rearing environment on the development of GABAA and central benzodiazepine receptor levels and novelty-induced fearfulness in the rat. Neuropsychopharmacol 22: 219–229.
Campbell S, Marriott M, Nahmias C, MacQueen GM (2004). Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry 161: 598–607.
Chalmers DT, Kwak SP, Mansour A, Akil H, Watson SJ (1993). Corticosteroids regulate brain hippocampal 5-HT1A receptor mRNA expression. J Neurosci 13: 914–923.
Chaouloff F, Berton O, Mormède P (1999). Serotonin and stress. Neuropsychopharmacology 21: S28–S32.
Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT (2001). Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 50: 260–265.
Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP et al (2005). Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci USA 102: 15653–15658.
Cohen RA, Grieve S, Hoth K, Paul RH, Sweet L, Tate D et al (2006). Early life stress and morphometry of the adult anterior cingulate cortex and caudate nuclei. Biol Psychiatry 59: 975–982.
Cowen PJ, Power AC, Ware CJ, Anderson IM (1994). 5-HT1A receptor sensitivity in major depression. A neuroendocrine study with buspirone. Br J Psychiatry 164: 372–379.
Czeh B, Lucassen P (2007). What causes the hippocampal volume decrease in depression? Eur Arch Psychiatry Clin Neurosci 257: 250–260.
Czesak M, Lemonde S, Peterson E, Rogaeva A, Albert PR (2006). Cell-specific repressor or enhancer activities of Deaf-1 at a serotonin 1A receptor gene polymorphism. J Neurosci 26: 1864–1871.
Deakin JFW (1998). The role of serotonin in depression and anxiety. Eur Psychiatry 13: 57S–63S.
DeBellis MD, Baum AS, Birmaher B, Keshavan MS, Eccard CH, Boring AM et al (1999). Developmental traumatology. Part 1: biological stress systems. Biol Psychiatry 45: 1259–1270.
De Gois S, Schafer MKH, Defamie N, Chen C, Ricci A, Weihe E et al (2005). Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci 25: 7121–7133.
Dettling AC, Feldon J, Pryce CR (2002a). Repeated parental deprivation in the infant common marmoset (Callithrix jacchus, primates) and analysis of its effects on early development. Biol Psychiatry 52: 1037–1046.
Dettling AC, Feldon J, Pryce CR (2002b). Early deprivation and behavioral and physiological responses to social separation/novelty in the marmoset. Pharmacol Biochem Behav 73: 259–269.
Dettling AC, Schnell CR, Maier C, Feldon J, Pryce CR (2007). Behavioral and physiological effects of an infant-neglect manipulation in a bi-parental, twinning primate: impact is dependent on familial factors. Psychoneuroendocrinology 32: 331–349.
Drevets WC, Frank E, Price JC, Kupfer DJ, Holt D, Greer PJ et al (1999). PET imaging of serotonin 1A receptor binding in depression. Biol Psychiatry 46: 1375–1387.
Duman RS, Heninger GR, Nestler EJ (1997). A molecular and cellular theory of depression. Arch Gen Psychiatry 54: 597–606.
Duman RS, Monteggia LM (2006). A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59: 1116–1127.
Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN (2003). Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 60: 804–815.
Eastwood SL, Harrison PJ (1998). Hippocampal and cortical growth-associated protein-43 messenger RNA in schizophrenia. Neuroscience 86: 437–448.
Eastwood SL, Harrison PJ (2000). Hippocampal synaptic pathology in schizophrenia, bipolar disorder and major depression: a study of complexin mRNAs. Mol Psychiatry 5: 425–432.
Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A et al (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112: 257–269.
Faraday MM (2002). Rat sex and strain differences in responses to stress. Physiol Behav 75: 507–522.
Fatemi SH, Earle JA, Stary JM, Lee S, Sedgewick J (2001). Altered levels of the synaptosomal associated protein SNAP-25 in hippocampus of subjects with mood disorders and schizophrenia. Neuroreport 12: 3257–3262.
Frey BN, Andreazza AC, Nery FG, Martins MR, Quevedo J, Soares JC et al (2007). The role of hippocampus in the pathophysiology of bipolar disorder. Behav Pharmacol 18: 419–430.
Frodl T, Schuele C, Schmitt G, Born C, Baghai T, Zill P et al (2007). Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch Gen Psychiatry 64: 410–416.
Fuchs E, Uno H, Flugge G (1995). Chronic psychosocial stress induces morphological alterations in hippocampal pyramidal neurons of the tree shrew. Brain Res 673: 275–282.
Gartside SE, Johnson DA, Leitch MM, Troakes C, Ingram CD (2003). Early life adversity programs changes in central 5-HT neuronal function in adulthood. Eur J Neurosci 17: 2401–2408.
Geuze E, Vermetten E, Bremner JD (2005). MR-based in vivo hippocampal volumetrics: 2. Findings in neuropsychiatric disorders. Mol Psychiatry 10: 160–184.
Greisen MH, Altar CA, Bolwig TG, Whitehead R, Wörtwein G (2005). Increased adult hippocampal brain-derived neurotrophic factor and normal levels of neurogenesis in maternal separation rats. J Neurosci Res 79: 772–778.
Griffin III WC, Skinner HD, Birkle DL (2005). Prenatal stress influences 8-OH-DPAT modulated startle responding and [3H]-8-OH-DPAT binding in rats. Pharmacol Biochem Behav 81: 601–607.
Gunnar M, Quevedo K (2007). The neurobiology of stress and development. Annu Rev Psychol 58: 145–173.
Groves JO (2007). Is it time to reassess the BDNF hypothesis of depression? Mol Psychiatry 12: 1079–1088.
Harris T, Brown GW, Bifulco A (1986). Loss of parent in childhood and adult psychiatric disorder: the role of lack of adequate parental care. Psychol Med 16: 641–659.
Harrison PJ (2002). The neuropathology of primary mood disorder. Brain 125: 1428–1449.
Harrison PJ, Eastwood SL (2001). Neuropathological studies of synaptic connectivity in the hippocampal formation in schizophrenia. Hippocampus 11: 508–519.
Hashimoto K, Shimizu E, Iyo M (2004). Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res Rev 45: 104–114.
Hashimoto K, Sawa A, Iyo M (2007). Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry 62: 1310–1316.
Heim C, Nemeroff CB (2001). The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry 49: 1023–1039.
Higo N, Oishi T, Yamashita A, Matsuda K, Hayashi M (1998). Gene expression of growth-associated proteins, GAP-43 and SCG10, in the hippocampal formation of the macaque monkey: nonradioactive in situ hybridization study. Hippocampus 8: 533–547.
Hirvonen J, Karlsson H, Kajander J, Lepola A, Markkula J, Rasi-Hakala H et al (2008). Decreased brain serotonin 5-HT1A receptor availability in medication-naive patients with major depressive disorder: an in-vivo imaging study using PET and [carbonyl-11C]WAY-100635. Int J Neuropsychopharmacol 11: 465–476.
Holderbach R, Clark K, Moreau JL, Bischofberger J, Normann C (2007). Enhanced long-term synaptic depression in an animal model of depression. Biol Psychiatry 62: 92–100.
Homberg JR, Schiepers OJG, Schoffelmeer ANM, Cuppen E, Vanderschuren LJMJ (2007). Acute and constitutive increases in serotonin levels reduce social play behaviour in peri-adolescent rats. Psychopharmacology 195: 175–182.
Hsu FC, Zhang GJ, Raol YSH, Valentino RJ, Coulter DA, Brooks-Kayal AR (2003). Repeated neonatal handling with maternal separation permanently alters hippocampal GABAA receptors and behavioral stress responses. Proc Natl Acad Sci USA 100: 12213–12218.
Huang EJ, Reichardt LF (2001). Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677–736.
Ichise M, Vines DC, Gura T, Anderson GM, Suomi SJ, Higley JD et al (2006). Effects of early life stress on [11C] DASB positron emission tomography imaging of serotonin transporters in adolescent peer- and mother-reared rhesus monkeys. J Neurosci 26: 4638–4643.
Jin H, Wu H, Osterhaus G, Wei J, Davis K, Sha D et al (2003). Demonstration of functional coupling between gamma-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc Natl Acad Sci USA 100: 4293–4298.
Jokela M, Keltikangas-Jarvinen L, Kivimaki M, Puttonen S, Elovainio M, Rontu R et al (2007). Serotonin receptor 2A gene and the influence of childhood maternal nurturance on adulthood depressive symptoms. Arch Gen Psychiatry 64: 356–360.
Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R (2005). Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Mol Brain Res 136: 29–37.
Kendler KS, Kessler RC, Neale MC, Heath AC, Eaves LJ (1993). The prediction of major depression in women: toward an integrated etiologic model. Am J Psychiatry 150: 1139–1148.
Kendler KS, Gatz M, Gardner CO, Pedersen NL (2006). A Swedish national twin study of lifetime major depression. Am J Psychiatry 163: 109–114.
Knable MB, Barci BM, Webster MJ, Meador-Woodruff J, Torrey EF (2004). Molecular abnormalities of the hippocampus in severe psychiatric illness: postmortem findings from the Stanley Neuropathology Consortium. Mol Psychiatry 9: 609–620.
Law AJ, Weickert CS, Webster MJ, Herman MM, Kleinman JE, Harrison PJ (2003). Expression of NMDA receptor NR1, NR2A and NR2B subunit mRNAs during development of the human hippocampal formation. Eur J Neurosci 18: 1197–1205.
Law AJ, Weickert CS, Hyde TM, Kleinman JE, Harrison PJ (2004). Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines. Am J Psychiatry 161: 1848–1855.
Le Corre SL, Sharp T, Young AH, Harrison PJ (1997). Increase of 5-HT7 (serotonin-7) and 5-HT1A (serotonin-1A) receptor mRNA expression in rat hippocampus after adrenalectomy. Psychopharmacol 130: 368–374.
Lenze S, Xiong C, Sheline YI (2008). Childhood adversity predicts earlier onset of major depression but not reduced hippocampal volume. Psychiatr Res Neuroimaging 162: 39–49.
Lippmann M, Bress A, Nemeroff CB, Plotsky PM, Monteggia LM (2007). Long-term behavioural and molecular alterations associated with maternal separation in rats. Eur J Neurosci 25: 3091–3098.
Liu D, Diorio J, Day JC, Francis DD, Meaney MJ (2000). Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci 3: 799–806.
Lopez JF, Chalmers DT, Little KY, Watson SJ (1998). Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol Psychiatry 43: 547–573.
Lopez-Figueroa AL, Norton CS, Lopez-Figueroa MO, Mellini-Dodel D, Burke S, Akil H et al (2004). Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biol Psychiatry 55: 225–233.
Lustig RH, Hua P, Wilson MC, Federoff HJ (1993). Ontogeny, sex dimorphism, and neonatal sex hormone determination of synapse-associated messenger RNAs in rat brain. Mol Brain Res 20: 101–110.
Manji HK, Drevets WC, Charney DS (2001). The cellular neurobiology of depression. Nat Med 7: 541–547.
Mann JJ (1999). Role of the serotonergic system in the pathogenesis of major depression and suicidal behavior. Neuropsychopharmacology 21: S99–S105.
McCullumsmith RE, Meador-Woodruff JH (2003). Expression of vesicular glutamate transporters one and two in medial temporal lobe structures in schizophrenia, bipolar disorder, and major depressive disorder [Abstract]. Schizophr Res 60: 65.
McEwen BS (1999). Stress and hippocampal plasticity. Annu Rev Neurosci 22: 105–122.
McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM (1997). Identification and characterization of the vesicular GABA transporter. Nature 389: 870–876.
McNamara RK, Namgung U, Routtenberg A (1996). Distinctions between hippocampus of mouse and rat: protein F1/GAP-43 gene expression, promoter activity, and spatial memory. Mol Brain Res 40: 177–187.
Meijer OC, van Oosten RV, de Kloet ER (1997). Elevated basal trough levels of corticosterone suppress hippocampal 5-hydroxytryptamine1A receptor expression in adrenally intact rats: implications for the pathogenesis of depression. Neuroscience 80: 419–426.
Meijer OC, Williamson A, Dallman MF, Pearce D (2000). Transcriptional repression of the 5-HT1A receptor promoter by corticosterone via mineralocorticoid receptors depends on the cellular context. J Neuroendocrinol 12: 245–254.
Montgomery AJ, Bench CJ, Young AH, Hammers A, Gunn RN, Bhagwagar Z et al (2001). PET measurement of the influence of corticosteroids on serotonin-1A receptor number. Biol Psychiatry 50: 668–676.
Nair A, Vadodaria KC, Banerjee SB, Benekareddy M, Dias BG, Duman RS et al (2006). Stressor-specific regulation of distinct brain-derived neurotrophic factor transcripts and cyclic AMP response element-binding protein expression in the postnatal and adult rat hippocampus. Neuropsychopharmacology 32: 1504–1519.
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002). Neurobiology of depression. Neuron 34: 13–25.
Neumaier JF, Sexton TJ, Hamblin MW, Beck SG (2000). Corticosteroids regulate 5-HT1A but not 5-HT1B receptor mRNA in rat hippocampus. Mol Brain Res 82: 65–73.
Neumaier JF, Edwards E, Plotsky PM (2002). 5-HT1B mRNA regulation in two animal models of altered stress reactivity. Biol Psychiatry 51: 902–908.
Parsey RV, Oquendo MA, Ogden RT, Olvet DM, Simpson N, Huang Y et al (2006). Altered serotonin 1A binding in major depression: A [carbonyl-C-11]WAY100635 positron emission tomography study. Biol Psychiatry 59: 106–113.
Pickering C, Gustafsson L, Cebere A, Nylander I, Liljequist S (2006). Repeated maternal separation of male Wistar rats alters glutamate receptor expression in the hippocampus but not the prefrontal cortext. Brain Res 1099: 101–108.
Pittenger C, Duman RS (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33: 88–109.
Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T (2007). Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90: 397–406.
Pryce CR, Dettling AC, Spengler M, Schnell CR, Feldon J (2004a). Deprivation of parenting disrupts development of homeostatic and reward systems in marmoset monkey offspring. Biol Psychiatry 56: 72–79.
Pryce CR, Dettling AC, Spengler M, Spaete C, Feldon J (2004b). Evidence for altered monoamine activity and emotional and cognitive disturbance in marmoset monkeys exposed to early life stress. Ann NY Acad Sci 1032: 245–249.
Pryce CR, Ruedi-Bettschen D, Dettling AC, Weston A, Russig H, Ferger B et al (2005a). Long-term effects of early-life environmental manipulations in rodents and primates: potential animal models in depression research. Neurosci Biobehav Rev 29: 649–674.
Pryce CR, Feldon J, Fuchs E, Knuesel I, Oertle T, Sengstag C et al (2005b). Postnatal ontogeny of hippocampal expression of the mineralocorticoid and glucocorticoid receptors in the common marmoset monkey. Eur J Neurosci 21: 1521–1535.
Reines A, Cereseto M, Ferrero A, Sifonios L, Podesta MF, Wikinski S (2008). Maintenance treatment with fluoxetine is necessary to sustain normal levels of synaptic markers in an experimental model of depression: correlation with behavioral response. Neuropsychopharmacology AOP (17 Oct 2007), DOI: 10.1038/sj.npp.1301596.
Roceri M, Cirulli F, Pessina C, Peretto P, Racagni G, Riva MA (2004). Postnatal repeated maternal deprivation produces age-dependent changes of brain-derived neurotrophic factor expression in selected rat brain regions. Biol Psychiatry 55: 708–714.
Roceri M, Hendriks W, Racagni G, Ellenbroek B, Riva MA (2002). Early maternal deprivation reduces the expression of BDNF and NMDA receptor subunits in rat hippocampus. Mol Psychiatry 7: 609–616.
Rosoklija G, Toomayan G, Ellis SP, Keilp J, Mann JJ, Latov N et al (2000). Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders. Arch Gen Psychiatry 57: 349–356.
Sabatini MJ, Ebert P, Lewis DA, Levitt P, Cameron JL, Mirnics K (2007). Amygdala gene expression correlates of social behavior in monkeys experiencing maternal separation. J Neurosci 27: 3295–3304.
Sahay A, Hen R (2007). Adult hippocampal neurogenesis in depression. Nat Neurosci 10: 1110–1115.
Sala M, Perez J, Soloff P, Ucelli di Nemi S, Caverzasi E, Soares JC et al (2004). Stress and hippocampal abnormalities in psychiatric disorders. Eur Neuropsychopharmacol 14: 393–405.
Sanchez MM, Ladd CO, Plotsky PM (2001). Early adverse experience as a developmental risk factor for later psychopathology: evidence from rodent and primate models. Dev Psychopathol 13: 419–449.
Sapolsky RM (2000). Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 57: 925–935.
Sargent PA, Kjaer KH, Bench CJ, Rabiner EA, Messa C, Meyer J et al (2000). Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635—effects of depression and antidepressant treatment. Arch Gen Psychiatry 57: 174–180.
Sequeira A, Klempan T, Canetti L, Ffrench-Mullen J, Benkelfat C, Rouleau GA et al (2007). Patterns of gene expression in the limbic system of suicides with and without major depression. Mol Psychiatry 12: 640–655.
Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW (1996). Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 93: 3908–3913.
Shively CA, Friedman DP, Gage HD, Bounds MC, Brown-Proctor C, Blair JB et al (2006). Behavioral depression and positron emission tomography-determined serotonin 1A receptor binding potential in cynomolgus monkeys. Arch Gen Psychiatry 63: 396–403.
Sibille E, Hen R (2001). Serotonin1A receptors in mood disorders: a combined genetic and genomic approach. Behav Pharmacol 12: 429–438.
Stamatakis A, Mantelas A, Papaioannou A, Pondiki S, Fameli M, Stylianopoulou F (2006). Effect of neonatal handling on serotonin 1A sub-type receptors in the rat hippocampus. Neuroscience 140: 1–11.
Stephan H, Baron G, Schwerdtfeger WK (1980). The Brain of the Common Marmoset (Callithrix jacchus): A Sterotaxic Atlas. Springer-Verlag: Berlin.
Stockmeier CA (2003). Involvement of serotonin in depression: evidence from postmortem and imaging studies of serotonin receptors and the serotonin transporter. J Psychiatr Res 37: 357–373.
Strobel A, Gutknecht L, Rothe C, Reif A, Mössner R, Zeng Y et al (2003). Allelic variation in 5-HT 1A receptor expression is associated with anxiety- and depression-related personality traits. J Neural Transm 110: 1445–1453.
Taylor M, Bhagwagar Z, Cowen PJ, Sharp T (2003). GABA and mood disorders. Psychol Med 33: 387–393.
Tian SY, Wang JF, Bezchlibnyk YB, Young LT (2007). Immunoreactivity of 43 kDa growth-associated protein is decreased in post mortem hippocampus of bipolar disorder and schizophrenia. Neurosci Lett 411: 123–127.
Tordera RM, Pei Q, Sharp T (2005). Evidence for increased expression of the vesicular glutamate transporter, VGLUT1, by a course of antidepressant treatment. J Neurochem 94: 875–883.
Van Riel E, Van Gemert NG, Meijer OC, Joëls M (2004). Effect of early life stress on serotonin responses in the hippocampus of young adult rats. Synapse 53: 11–19.
Vawter MP, Thatcher L, Usen N, Hyde TM, Kleinman JE, Freed WJ (2002). Reduction of synapsin in the hippocampus of patients with bipolar disorder and schizophrenia. Mol Psychiatry 7: 571–579.
Vythilingam M, Heim C, Newport J, Miller AH, Anderson E, Bronen R et al (2002). Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry 159: 2072–2080.
Walker MA, Highley JR, Esiri MM, McDonald B, Roberts HC, Evans SP et al (2002). Estimated neuronal populations and volumes of the hippocampus and its subfields in schizophrenia. Am J Psychiatry 159: 821–828.
Webster MJ, Knable MB, O’Grady J, Orthmann J, Weickert CS (2002). Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol Psychiatry 7: 985–994.
Webster MJ, Weickert CS, Herman M, Hyde T, Kleinman JE (2001). Synaptophysin and GAP-43 mRNA levels in the hippocampus of subjects with schizophrenia. Schizophr Res 49: 89–98.
Weickert CS, Hyde TM, Lipska BK, Herman MM, Weinberger DR, Kleinman JE (2003). Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry 8: 592–610.
Whitaker-Azmitia PM (2001). Serotonin and brain development: role in human developmental diseases. Brain Res Bull 56: 479–485.
Whitfield Jr HJ, Brady LS, Smith MA, Mamalaki E, Fox RJ, Herkenham M (1990). Optimization of cRNA probe in situ hybridization methodology for localization of glucocorticoid receptor mRNA in rat brain: a detailed protocol. Cell Mol Neurobiol 10: 145–157.
Wilson NR, Kang JS, Hueske EV, Leung T, Varoqui H, Murnick JG et al (2005). Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci 25: 6221–6234.
Yildiz-Yesiloglu A, Ankerst DP (2006). Review of 1H magnetic resonance spectroscopy findings in major depressive disorder: a meta-analysis. Psychiatr Res Neuroimaging 147: 1–25.
Zhang L, Ma W, Barker J, Rubinow DR (1999). Sex differences in expression of serotonin receptors (subtypes 1A and 2A) in rat brain: a possible role of testosterone. Neuroscience 94: 251–259.
Acknowledgements
The study was funded by the Wellcome Trust (grant no. 068856), and the National Science Foundation, Switzerland (Project grant no. 3167791.02) and National Center for Competence in Research: Swiss Etiological Study of Adjustment and Mental Health (grant no. 51A240–104890). We thank Phil Burnet, Andrea Dettling and Sharon Eastwood for their contributions and Phil Cowen for helpful comments on the paper. Valerie West kindly prepared the reference list.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
In the last 3 years PJH has received unrestricted educational grants from GlaxoSmithKline (GSK), and honoraria for educational lectures or chairing scientific meetings from Bristol Myers Squibb, GSK, Janssen, Lilly, Merck, Sanofi and Servier, and has been a scientific advisor to Curidium, Janssen, Merck, and Wyeth. None of the other authors have any disclosures.
Rights and permissions
About this article
Cite this article
Law, A., Pei, Q., Walker, M. et al. Early Parental Deprivation in the Marmoset Monkey Produces Long-Term Changes in Hippocampal Expression of Genes Involved in Synaptic Plasticity and Implicated in Mood Disorder. Neuropsychopharmacol 34, 1381–1394 (2009). https://doi.org/10.1038/npp.2008.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2008.106
Keywords
This article is cited by
-
Transgenerational disruption of functional 5-HT1AR-induced connectivity in the adult mouse brain by traumatic stress in early life
Molecular Psychiatry (2017)
-
Identification of dorsal–ventral hippocampal differentiation in neonatal rats
Brain Structure and Function (2015)
-
Developmental exposure to SSRIs, in addition to maternal stress, has long-term sex-dependent effects on hippocampal plasticity
Psychopharmacology (2015)
-
Meta-analysis of diffusion tensor imaging studies shows altered fractional anisotropy occurring in distinct brain areas in association with depression
Biology of Mood & Anxiety Disorders (2011)
