
Abstract
Competition between electron localization and delocalization in Mott insulators underpins the physics of strongly correlated electron systems. Photoexcitation, which redistributes charge, can control this many-body process on the ultrafast timescale1,2. So far, time-resolved studies have been carried out in solids in which other degrees of freedom, such as lattice, spin or orbital excitations3,4,5, dominate. However, the underlying quantum dynamics of ‘bare’ electronic excitations has remained out of reach. Quantum many-body dynamics are observed only in the controlled environment of optical lattices6,7 where the dynamics are slower and lattice excitations are absent. By using nearly single-cycle near-infrared pulses, we have measured coherent electronic excitations in the organic salt ET-F2TCNQ, a prototypical one-dimensional Mott insulator. After photoexcitation, a new resonance appears, which oscillates at 25 THz. Time-dependent simulations of the Mott–Hubbard Hamiltonian reproduce the oscillations, showing that electronic delocalization occurs through quantum interference between bound and ionized holon–doublon pairs.
Similar content being viewed by others
Main
In Mott insulators, conductivity at low energies is prevented by the repulsion between electrons. This state is fundamentally different from that of conventional band insulators, in which Bragg scattering from the lattice opens gaps in the single-particle density of states. The electronic structure of Mott insulators is, therefore, sensitive to doping. Photoexcitation, in analogy to static doping, can trigger large changes in the macroscopic properties8. However, the coherent physics driving these transitions has not been fully observed because the many-body electronic dynamics are determined by hopping and correlation processes that persist for only a few femtoseconds.
We report measurements of coherent many-body dynamics with ultrafast optical spectroscopy in the one-dimensional Mott insulator bis(ethylendithyo)-tetrathiafulvalene-difluorotetracyanoquinodimethane (ET-F2TCNQ). Several factors make this possible: ET-F2TCNQ has a narrow bandwidth (∼100 meV), which corresponds to hopping times of tens of femtoseconds; the material has a weak electron–lattice interaction; we use a new optical device producing pulses of 9 fs at the 1.7 μm Mott gap; we study this physics in a one-dimensional system, allowing the evolution of the many-body wavefunction to be calculated and compared with experimental data.
ET-F2TCNQ is a stacked molecular ionic solid9 in which ET molecules are donors and F2TCNQ are acceptors, which form quasi one-dimensional chains of ET molecules. Conducting electrons are localized on ET sites because the large onsite Coulomb repulsion (U∼1 eV) exceeds the single-electron hopping amplitude (t∼0.1 eV; ref. 10).
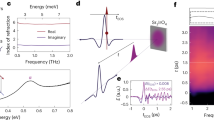
Figure 1 shows the optical properties of ET-F2TCNQ. No Drude weight is found at low energies and light polarized parallel to the ET chains is strongly reflected at ∼0.7 eV. This feature corresponds to intersite charge transfer between neighbouring ET ions, resulting in a hole on one lattice site (a holon) and a neighbouring site with two electrons (a doublon). The charge transfer feature in ET-F2TCNQ is sharp, reflecting a bandwidth lower than the gap energy. Figure 1a shows the static reflectivity of ET-F2TCNQ fitted with a multi-Lorentzian dielectric function of the form  , where Aj is the oscillator strength, ωj0 is the resonance frequency and γj is the damping rate. A single oscillator at ωMott=0.675 eV is sufficient to describe the Mott gap. Figure 1b shows the optical conductivity obtained from the imaginary part of the fitted dielectric function.
, where Aj is the oscillator strength, ωj0 is the resonance frequency and γj is the damping rate. A single oscillator at ωMott=0.675 eV is sufficient to describe the Mott gap. Figure 1b shows the optical conductivity obtained from the imaginary part of the fitted dielectric function.

a, Measured reflectivity (red line) and modelled reflectivity (black line). The reflectivity was obtained from the dielectric function described in the text with  . b, Optical conductivity extracted from the imaginary part of the dielectric function.
. b, Optical conductivity extracted from the imaginary part of the dielectric function.
Photoexcitation across the Mott gap of ET-F2TCNQ transiently generates a metallic state11, evidenced by the transfer of spectral weight from the Mott gap to a Drude response at low frequencies resulting from charge delocalization. To observe the initial delocalization dynamics, short light pulses are needed. Even for the low hopping amplitudes of this compound (t∼100 meV) we expect electron delocalization to require a time of order h/t∼40 fs, where h is Planck’s constant. Similarly, we expect correlated electrons to be dressed on timescales of the order of h/U. Furthermore, the pulses need to be resonant with the 0.7 eV charge transfer resonance, corresponding to a wavelength of 1.7 μm, where sub-10-fs pulses have not been previously achieved. Therefore, we developed a source at 1.7 μm with 9 fs duration12.
Figure 2 reports time-resolved reflectivity measurements of the photoinduced response of ET-F2TCNQ in the paramagnetic, Mott insulating phase at room temperature. The time-dependent reflectivity was probed with a replica of the pump pulse and spectrally resolved. Both pump and probe were polarized along the crystal’s a axis, to excite charges across the Mott gap. Figure 2a shows the spectrally integrated reflectivity change. After a prompt decrease, relaxation back to the ground state occurs with a bi-exponential decay with time constants of 130 fs and 840 fs. Excitation with light perpendicular to the chains produced no observable dynamics, confirming that the experiments address the one-dimensional Mott physics of ET-F2TCNQ.

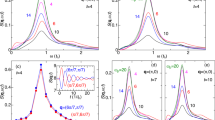
a, Spectrally integrated, time-dependent optical reflectivity of ET-F2TCNQ with a bi-exponential fit (dashed red curve). The resonance at the Mott gap collapses and recovers with two time constants of 130 fs and 840 fs. b, Two-dimensional colour plot of the time-dependent reflectivity. The lineouts show the evolution of the reflectivity (red line) together with the fitted reflectivity (dashed black line) compared to the static reflectivity (thin black line). The shaded lines represent the contribution to the reflectivity from the Lorentzian functions used to fit the peaks. c, Normalized reflectivity at early times, showing oscillations on the red edge of the spectrum. The right-hand side panel shows a Fourier transform of the oscillations in the reflectivity at 2 μm.
Figure 2b shows the spectrally resolved reflectivity. A prompt shift of spectral weight towards lower energies is followed by a drop in the reflectivity at the Mott gap. The dashed lines in Fig. 2 show the fitted reflectivity at each delay. Whereas the unperturbed reflectivity was accurately described by a single oscillator at the Mott gap, the transient reflectivity of the photoexcited system requires an additional, lower energy, oscillator. The appearance of a new resonance, which is clearly visible in the reflectivity lineouts of Fig. 2, suggests that below-gap bound states are formed, reminiscent of the bound holon–doublon pairs observed in other Mott insulators13.
The characteristics of this new peak are time-dependent, as shown in Fig. 2c, where we have normalized the reflectivity at each time step. Two contours are shown in Fig. 2c. On the blue side, a prompt redshift and recovery of the resonance is observed, whereas the red side shows a longer-lived component, containing a damped oscillatory response at 25 THz. Static Raman data on ET-F2TCNQ (see Supplementary Information) do not show any equivalent features, strongly suggesting that the oscillation is not due to coherent phonons, but of an electronic origin.
To investigate such dynamics, we used a one-dimensional Mott–Hubbard Hamiltonian for a half-filled chain, with N=10 sites, with electron hopping, t, and onsite and nearest-neighbour Coulomb repulsion U and V,

where cl,σ† and cl,σ are the creation and annihilation operators for an electron at site l with spin σ,nl,σ is the number operator and nl=nl,↑+nl,↓. We described the initial state as  , where
, where  represents a many-body wavefunction with one electron per site and total spin-vector
represents a many-body wavefunction with one electron per site and total spin-vector  . This reflects the fact that, at room temperature, charges are localized, but possess no magnetic ordering.
. This reflects the fact that, at room temperature, charges are localized, but possess no magnetic ordering.
We calculate the static optical conductivity (see the Methods section) to find values of U,V and t that provide the best fit to the static measurements. The best fit, shown in Fig. 3c (t=−200 fs), gave U=820 meV, V =100 meV and t=50 meV. It was not possible to fit the optical conductivity using U and t alone and the intersite correlation energy, V, was needed14.

a, Two-dimensional colour plot of the time- and wavelength-dependent optical conductivity extracted from the reflectivity data. b, Numerical simulation of the optical conductivity with σ(ω,T)=pg(T)σg(ω)+pe(T)σe,1(ω). c, Comparison of the numerical model to the extracted optical conductivity. The model is in increasing agreement with measurements for time delays greater than 200 fs, indicating that the recovery of the system is due to the decay of holon–doublon pairs.
These static parameters were used to fit to the time-dependent optical properties. We considered states created by the laser, ρe,L, which consist of a neighbouring holon–doublon pair, delocalized over L lattice sites, with an optical conductivity σe,L(ω). To model the time-dependence, the system was described by an incoherent mixture of two states: ρ(T)=pg(T)ρg+pe(T)ρe,1, where T is pump–probe time delay and ps(T) is the fraction of the sample in state s. The time-dependent optical conductivity was described as σ(ω,T)=pg(T)σg(ω)+pe(T)σe,1(ω), pg(T) and pe(T) were varied, under the constraint pg(T)+pe(T)=1, to obtain the best fit.
The result is shown in Fig. 3b, where the calculated time-dependent optical conductivity is compared to that obtained from the transient reflectivity by fitting the dielectric function with two time-dependent oscillators. The model provides a good fit at long time delays (>200 fs), indicating that the long-term dynamics are dictated by incoherent holon–doublon decay. However, the model fails at early times when coherent processes are primarily responsible for the dynamics.
To simulate the coherent dynamics, we calculate the time evolution of the state ρe,10 under the Hamiltonian. The temporal evolution has no free parameters, and the optical conductivity was calculated after the state evolved for a time T. In Fig. 4 the normalized result of this simulation is compared to the normalized experimental optical conductivity. The simulation reproduces the 25 THz oscillations observed experimentally, as shown by the Fourier transforms in Fig. 4c. The numerical simulation contains no damping or dephasing, resulting in a narrower Fourier transform, yet, it is remarkable that this model reproduces the frequency with this accuracy.

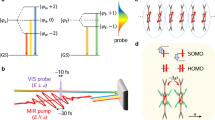
a, The retrieved normalized optical conductivity during the first 150 fs, with oscillations in spectral weight with a period of approximately 40 fs. b, Time-dependent simulation of the quantum evolution of the optical conductivity under the Mott–Hubbard Hamiltonian showing similar oscillations. c, Fourier transforms of the oscillations in a and b, showing an excellent agreement at ∼25 THz. d, Representations of the initial state, excited holon–doublon bound state and ionized holon–doublon state. Right: Mapping the bound and ionized holon–doublon states onto two potential energy wells offset by V and coupled by tunnelling amplitude t.
The origin of these oscillations can be understood by considering interference between the different photoexcitation paths of the Mott insulator. Optical excitation acts on the ground state |…111111…〉 to create bound states of the form |…110211…〉. In this case, the holon–doublon pair is bound by an energy V. This state can evolve into a superposition of bound states and ionized states of the form |…110121…〉 by electron hopping. These excitations interfere in the time domain, giving rise to the observed oscillations. This conclusion is validated by simulations with V =0, in which these oscillations are not observed (see Supplementary Information).
This physics is reminiscent of single-particle excitations in multiple quantum wells. An electron is transferred from a well of depth U into a neighbouring well with depth V, representing the bound holon–doublon pair. This ‘exciton’ can tunnel, with an energy t, into a third well, representing the ionized pair (see Fig. 4d). Such a system will oscillate at  , in close agreement with the experimental and numerical observations. Thus, our experiments highlight, in a room-temperature solid, coherent many-body physics that has, so far, been accessible only in ultracold gases15. Our experiments captured dynamics on the timescale associated with hopping and intersite correlations. However, higher temporal resolution could observe dressing resulting from onsite Coulomb correlations, and the coherent formation of the holon–doublon pairs. These measurements would require temporal resolutions approaching the attosecond regime16 and, therefore, we anticipate that time-resolved photoelectron spectroscopy17 and ultrafast soft X-ray techniques18 will play major roles in these studies.
, in close agreement with the experimental and numerical observations. Thus, our experiments highlight, in a room-temperature solid, coherent many-body physics that has, so far, been accessible only in ultracold gases15. Our experiments captured dynamics on the timescale associated with hopping and intersite correlations. However, higher temporal resolution could observe dressing resulting from onsite Coulomb correlations, and the coherent formation of the holon–doublon pairs. These measurements would require temporal resolutions approaching the attosecond regime16 and, therefore, we anticipate that time-resolved photoelectron spectroscopy17 and ultrafast soft X-ray techniques18 will play major roles in these studies.
Methods
Sample preparation.
Single crystals were grown by first purifying commercially available ET molecules and synthesizing F2TCNQ (ref. 19) by repeated cycles of recrystallization and sublimation. Single crystals of approximately 3 mm×10 mm×0.5 mm were achieved by slowly cooling a hot chlorobenzene solution of purified ET and F2TCNQ.
Experimental set-up.
A near-infrared optical parametric amplifier is driven by an amplified Ti:sapphire laser (100 μJ, 150 fs, 1 kHz pulses at 800 nm). A white-light seed, generated in a sapphire plate, is amplified in a 3 mm Type-I β-barium-borate crystal to ≈2 μJ energy. The ultrabroadband optical parametric amplifier pulses, with a spectrum covering the 1,200–2,200 nm range, are compressed using a deformable mirror to a nearly transform-limited 9 fs duration. The pump fluence at the sample surface was 3.5 mJ cm−2. Transient reflectivity is measured in a degenerate pump–probe configuration at a near-normal angle of incidence and the probe spectrum is detected by an InGaAs optical multichannel analyser.
Numerical simulation.
The initial state of the system is assumed to be described by the density matrix,  . The laser creates excited states of the form ρe,L∝XLρgXL†, where the excitation operator
. The laser creates excited states of the form ρe,L∝XLρgXL†, where the excitation operator  corresponds to the creation of a single holon–doublon pair at neighbouring sites, delocalized over L lattice sites.
corresponds to the creation of a single holon–doublon pair at neighbouring sites, delocalized over L lattice sites.
The optical conductivity of a given state, ρ, is calculated using the unequal time current–current correlation function cj j(τ,τ′)=tr[ρ j(τ)j(τ′)]θ(τ−τ′), where τ>τ′,  is the current density operator, τ is time, θ(τ) is the Heaviside function and j(τ)=exp(i H τ)jexp(−i H τ) is the current operator in the Heisenberg picture. The definition of H is given in the main text.
is the current density operator, τ is time, θ(τ) is the Heaviside function and j(τ)=exp(i H τ)jexp(−i H τ) is the current operator in the Heisenberg picture. The definition of H is given in the main text.
For the incoherent model we take cj j(τ,τ′)=cj j(τ,0)=cj j(τ) and the regular finite-frequency optical conductivity then follows from cj j(ω), the Fourier transform taken with respect to τ, as σ(ω>0)∝(Re{cj j(ω)})/ω. The total evolution time over which cj j(τ) is computed was limited to τmax=5h/t. The Fourier transform was carried out with a Gaussian windowing function exp[−4(τ/τmax)2], leading to a broadening and smoothing of cj j(ω) compared with its exact limit, which is instead composed of numerous δ-functions. This is justified because we focus on features in the high angular frequency range 8t<ω<32t and because the probe pulse used in the experiment has similar spectral limitations.
The time-dependent optical conductivity for the state ρe,10 was calculated using the full two-time current–current correlation function cj j(τ,T), where T is the pump–probe time delay and the Fourier transform is taken with respect to τ−T.
Change history
09 December 2010
In the version of this Letter originally published online, the x axis of Fig. 4a was incorrect. This has now been corrected in all versions of the Letter.
References
Zu, G. et al. Transient photoinduced conductivity in single crystals of YBa2Cu3O6.3: Photodoping to the metallic state. Phys. Rev. Lett. 67, 2581–2584 (1991).
Miyano, K., Tanaka, T., Tomioka, Y. & Tokura, Y. Photoinduced insulator-to-metal transition in a perovskite manganite. Phys. Rev. Lett. 78, 4257–4260 (1997).
Cavalleri, A. et al. Evidence for a structurally-driven insulator-to-metal transition in VO2: A view from the ultrafast timescale. Phys. Rev. B 70, 161102 (2004).
Wall, S., Prabhakaran, D., Boothroyd, A. T. & Cavalleri, A. Ultrafast coupling between light, coherent lattice vibrations, and the magnetic structure of semicovalent LaMnO3 . Phys. Rev. Lett. 103, 097402 (2009).
Polli, D. et al. Coherent orbital waves in the photo-induced insulator–metal dynamics of a magnetoresistive manganite. Nature Mater. 6, 643–647 (2007).
Greiner, M. et al. Quantum phase transition from a superfluid to a Mott insulator in a gas of ultracold atoms. Nature 415, 39–44 (2001).
Greiner, M., Mandel, O., Haensch, T. W. & Bloch, I. Collapse and revival of the matter wave field of a Bose–Einstein condensate. Nature 419, 51–54 (2002).
Koshihara, S. Photoinduced valence instability in the organic molecular compound tetrathiafulvalene-p-chloranil (TTF-CA). Phys. Rev. B. 42, 6853–6856 (1990).
Hasegawa, T. et al. Electronic states and anti-ferromagnetic order in mixed-stack charge-transfer compound (BEDT-TTF)(F2TCNQ). Solid State Commun. 103, 489–493 (1997).
Hasegawa, T. et al. Mixed-stack organic charge-transfer complexes with intercolumnar networks. Phys. Rev. B 62, 10059–10066 (2000).
Okamoto, H. et al. Photoinduced metallic state mediated by spin-charge separation in a one-dimensional organic Mott insulator. Phys. Rev. Lett 98, 037401 (2007).
Brida, D. et al. Sub-two-cycle light pulses at 1.6 μm from an optical parametric amplifier. Opt. Lett. 33, 741–743 (2008).
Kim, K. W., Gu, G. D., Homes, C. C. & Noh, T. W. Bound excitons in Sr2CuO3 . Phys. Rev. Lett. 101, 177404 (2008).
Gomi, H., Takahashi, A., Ueda, T., Itoh, H. & Aihara, M. Photogenerated holon–doublon cluster states in strongly correlated low-dimensional electron systems. Phys. Rev. B 71, 045129 (2005).
Jordens, R. et al. A Mott insulator of fermionic atoms in an optical lattice. Nature 455, 204–207 (2008).
Cavalieri, A. L. et al. Attosecond spectroscopy in condensed matter. Nature 449, 1029–1032 (2007).
Perfetti, L. et al. Time evolution of the electronic structure of 1 T-TaS2 through the insulator–metal transition. Phys. Rev. Lett. 97, 067402 (2006).
Cavalleri, A., Rini, M. & Schoenlein, R. W. Ultra-broadband femtosecond measurements of the photo-induced phase transition in VO2: From the Mid-IR to the hard X-rays. J. Phys. Soc. Jpn 75, 011004 (2006).
Uno, M. et al. A new route to phenylenedimalononitrile and the analogues using palladium-catalyzed carbon–carbon bond formation. Tetrahedron Lett. 26, 1553–1556 (1985).
Acknowledgements
This work was supported by the European Community Access to Research Infrastructure Action, Contract RII3-CT-2003-506350 (Centre for Ultrafast Science and Biomedical Optics, LASERLAB-EUROPE). S.R.C and D.J thank the National Research Foundation and the Ministry of Education of Singapore for support. A.A. is supported by the Royal Society. H.O. is grateful for support by a Grant-in-Aid for Scientific Research (No. 20110005) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Contributions
S.W., D.B., H.P.E. and G.C. carried out the pump–probe experiments. D.B., S.B. and G.C. designed and built the experimental apparatus. H.U., Y.T., T.H. and H.O. provided samples. S.W. analysed the experimental data. S.R.C. and D.J. carried out the numerical simulations. S.W., A.C., A.A., S.R.C. and D.J. interpreted the data and the simulations. A.C and S.W. wrote the manuscript. A.C. conceived and coordinated the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Information (PDF 287 kb)
Rights and permissions
About this article
Cite this article
Wall, S., Brida, D., Clark, S. et al. Quantum interference between charge excitation paths in a solid-state Mott insulator. Nature Phys 7, 114–118 (2011). https://doi.org/10.1038/nphys1831
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys1831
This article is cited by
-
A Hubbard exciton fluid in a photo-doped antiferromagnetic Mott insulator
Nature Physics (2023)
-
Axial Higgs mode detected by quantum pathway interference in RTe3
Nature (2022)
-
Measurement of 10 fs pulses across the entire Visible to Near-Infrared Spectral Range
Scientific Reports (2020)
-
Ultrafast evolution and transient phases of a prototype out-of-equilibrium Mott–Hubbard material
Nature Communications (2017)
-
Non-linear quantum-classical scheme to simulate non-equilibrium strongly correlated fermionic many-body dynamics
Scientific Reports (2016)
