
Abstract
Surface-enhanced Raman scattering (SERS)-based signal amplification and detection methods using plasmonic nanostructures have been widely investigated for imaging and sensing applications. However, SERS-based molecule detection strategies have not been practically useful because there is no straightforward method to synthesize and characterize highly sensitive SERS-active nanostructures with sufficiently high yield and efficiency, which results in an extremely low cross-section area in Raman sensing. Here, we report a high-yield synthetic method for SERS-active gold–silver core–shell nanodumbbells, where the gap between two nanoparticles and the Raman-dye position and environment can be engineered on the nanoscale. Atomic-force-microscope-correlated nano-Raman measurements of individual dumbbell structures demonstrate that Raman signals can be repeatedly detected from single-DNA-tethered nanodumbbells. These programmed nanostructure fabrication and single-DNA detection strategies open avenues for the high-yield synthesis of optically active smart nanoparticles and structurally reproducible nanostructure-based single-molecule detection and bioassays.
Similar content being viewed by others


Main
Extensive studies have been focused on unveiling the mechanism and robust evidence of single-molecule SERS phenomena1,2,3,4,5,6,7,8,9,10,11,12,13. SERS-active structures are generally geometry-controlled nanostructures14 and nanoparticle aggregates (often resulting from nonspecific aggregation) including dimeric structures15 in which the analyte is localized at a junction between the nanoparticles (‘hot spot’). However, SERS-based single-molecule detection generally faces a problem with structural reproducibility, as particle structure and interparticle distance can markedly affect Raman signals and constructing robust SERS-active nanostructures still remains challenging15,16,17,18. Recently, a quantitative measurement of the distribution of site enhancement in SERS was reported19. The hottest SERS-active sites (local enhancement factor is >109) accounted for only 63 sites in 1,000,000 of the total sites, but contributed 24% of the overall SERS intensity19. In these regards, assembling SERS-active nanoparticles into well-defined and reproducible hot SERS nanostructures and subsequent detection of Raman signals from individual SERS-active nanostructures would be greatly beneficial17,18. Owing to the programmability and structural plasticity of DNA, it was recently demonstrated as a powerful molecular tool for organizing nanostructures as one-dimensional groupings20, onto two-dimensional assemblies21,22,23 or into three-dimensional crystalline groupings24,25,26,27. However, DNA-directed nanoparticle assembly strategies for the synthesis of dimer or multimer nanostructures were based on electrophoretic separation28,29 or anionic exchange liquid chromatography30, both of which suffered low synthetic efficiency or limited scalability for specific nanostructures. Most recently, a magnetic-microparticle-based stepwise surface-encoding strategy for the assembly of nanoclusters was reported31. By using DNA-encoded nanoparticles and the steric hindrance of Au nanoparticles hybridized on magnetic particles to fabricate desired nanostructures in a stepwise manner, high-purity synthesis of symmetric dimeric nanostructures or janus nanoclusters was possible with a relatively high yield (∼73%).
Here, we report a high-yield synthetic strategy to obtain gap-tailorable gold–silver core–shell nanodumbbells (GSNDs) and subsequent hot SERS-based single-molecule detection with structurally reproducible dimetric nanostructures (Fig. 1). Using a stoichiometric control over the number of tethering DNA molecules on the Au nanoparticle surface and a subsequent magnetic-particle-based separation method, Au nanoparticle heterodimers were successfully synthesized in a relatively high yield by means of a single-target-DNA hybridization (Fig. 1a). Importantly, in this heterodimer, a single Raman-active Cy3 dye molecule is located between two DNA-tethered particles. In the next step, Ag shells were formed on the surface of the dimeric Au nanoparticles, and the Ag shell thickness was controlled on the nanometre scale to generate gap-engineerable32, DNA-embedded GSNDs (Fig. 1b). To detect a Raman signal from each single-DNA-captured GSND, atomic force microscope (AFM)-coupled nano-Raman spectroscopy was used. In this spectroscopic set-up, the laser focal spot was matched with the centre of the AFM tip for symmetrical scattering on the AFM tip end, which enables us to correlate the Raman signal with the AFM topographical image with an overlap precision of <100 nm (Fig. 1c). Along with a proper gap size between the core–shell nanoparticles, the size, shape and composition of the nanoparticles are also critical factors to obtain single-molecule sensitivity16. We found that optimized GSNDs have single-molecule sensitivity with high structural reproducibility from particle to particle.

a, A high-yield synthetic scheme for the Au nanoparticle heterodimers using stoichiometric DNA modification and magnetic purification. b, Nanometre-scale silver-shell growth-based gap-engineering in the formation of the SERS-active GSND. c, AFM-correlated nano-Raman spectroscopy set-up (laser focal diameter is 250 nm) for the detection of a Raman signal from a single GSND particle. More details about this set-up can be found in ref. 39.
In the synthesis of Raman-active GSNDs, first, Au nanoparticles were modified with two different kinds of DNA sequence (a protecting sequence and a target-capture sequence; Fig. 1a). The 20 nm Au nanoparticle probe A was functionalized with two kinds of 3′-thiol-modified DNA sequence, and the 30 nm Au nanoparticle probe B was functionalized by two kinds of 5′-thiol-modified DNA sequence (in this case, the target-capture sequence was modified with a Cy3 dye). The molar ratios of the two kinds of sequence were 99:1 (protecting sequence/target-capture sequence) for probe A and 199:1 for probe B. These ratios were used to modify ∼1 target-capture DNA per probe and based on nanoparticle size-dependent DNA loading capacity33. Importantly, the Raman-active Cy3 dye was pre-conjugated to the target-capture sequence (probe B alone) so that the dye could be located at the junction of the single-DNA-interconnected probes A and B (Fig. 1a,b). The number of Cy3 molecules released from 30 nm probe B was estimated by the fluorescence emission intensity (λex=520 nm). The number of Cy3-modified oligonucleotides per probe B is about 0.75, indicating that ∼75% of probe B particles have one Raman-active molecule per probe (Supplementary Fig. S1). Having ∼1 Raman dye per probe is important because our goal is to synthesize Au nanoparticle heterodimers in a high yield by means of single-target-DNA hybridization. Next, the oligonucleotide-modified probes A and B were captured by magnetic particles and purified by a magnetic-separation process to remove the probes without target-capture DNA strands. To prepare these magnetic particles, tosyl-modified magnetic particles (5 μl of 1.08 μm beads (100 mg ml−1, Invitrogen)) were functionalized by amine-modified target-DNA complements (25 nmol) for probes A and B, respectively (see the Methods section for sequence details). After repeated washing with a washing buffer (200 μl, 0.1 M sodium borate, pH 9.5) five times and phosphate-buffered saline (PBS) solution (100 μl, 10 mM phosphate, 0.3 M NaCl, pH 7.4) two times, the magnetic particles were allowed to react with the nanoparticle probes at room temperature for 1 h with vigorous vortexing. A magnetic field was applied for the separation of target-DNA complement-modified nanoparticle magnetic probes. The separated complexes were further washed with buffer solution (100 μl, 0.3 M PBS) three times to remove unhybridized, non-functionalized probes. After the nanoparticle probes were released from the magnetic beads at 65 ∘C (above the melting temperatures (Tm) of the probes: Tm (Probe A)=38 ∘C, Tm (Probe B)=40 ∘C) for 5 min, magnetic separation was carried out at the same temperature (65 ∘C, 2 min) in buffer solution (0.3 M PBS, 100 μl). The concentration of separated probes was determined by an ultraviolet–visible spectrometer. Finally, the separated 39.2 μl probes A (0.38 nM) and 82.7 μl probes B (0.18 nM) were hybridized with 5.3 μl of target DNA (11.3 μM) in 0.3 M PBS for 3 h at room temperature. The formation of Au nanoparticle dimers was verified by ultraviolet–visible spectroscopy (Fig. 2a) and high-resolution transmission electron microscope (HRTEM) images (Fig. 2a). The ultraviolet–visible spectra show a very small redshift and increased absorption around 600 nm after dimer formation, which is in good agreement with the previous results20,31,34. TEM images show synthesized Au nanoparticle dimers (Fig. 2a, top) and a DNA-tethered Au nanoparticle dimer with a nanogap (Fig. 2a, bottom). By a statistical analysis of 250 particles, we found that nearly 80% of the particles existed as a dimer, 10% of the particles as a monomer and <10% as a multimer (trimer, tetramer and so on) (Supplementary Fig. S2). Next, to confirm that the Cy3-modified DNA was located at the junction site between the DNA-tethered particles, the position of this Cy3 molecule in the dimer was verified by local electron energy-loss spectroscopy probing in the scanning transmission electron microscopy geometry (Supplementary Fig. S3). The subtle difference in the π* peak at ∼285 eV in the electron energy-loss spectrum between the junction of the nanoparticle pair and the background or other part of the nanoparticle pair suggests a relatively high abundance of carbon–carbon double bonds (C=C) from the Cy3 molecule near the junction area35. The interparticle distance from Au nanoparticle surface-to-surface (d1s−s) seems to be about 3–4 nm by HRTEM analysis, and the particle distance between Au nanoparticle core-to-core is likely to be ∼26 nm (d1c−c, Fig. 2a). However, this does not provide an accurate measurement for the interparticle distance because the distance between dried particles under vacuum should be shorter than that of particles in solution36. Finally, the DNA-tethered Au nanoparticle dimers were coated with silver by means of a nanometre-scale silver-shell deposition process on the Au nanoparticle surface to form the GSNDs. In a typical experiment, 250 μl of Au nanoparticle dimer solution was reacted with 1 mM AgNO3 solution at room temperature for 3 h in the presence of 100 μl of 1% poly-N-vinyl-2-pyrrolidone as a stabilizer and 50 μl of 0.1 M L-sodium ascorbate as a reductant in 0.3 M PBS solution. In this reaction, we confirmed that L-sodium ascorbate, a mild reducing agent, did not cause any noticeable damage to the Cy3 molecules conjugated to the oligonucleotides, as shown in Supplementary Fig. S4. The Ag shell thicknesses of the Au–Ag core–shell particles were ∼3 nm, ∼5 nm and ∼10 nm when using 30 μl, 40 μl and 70 μl of 1 mM AgNO3 solution, respectively (Fig. 2c-2, 2c-3 and 2c-4). Under the same conditions used for the GSNDs, Au–Ag core–shell monomers with ∼5 nm and ∼10 nm Ag shell thicknesses were also synthesized to be compared with Au nanoparticle probe B (the Ag shell thickness is 5 nm for Fig. 2c-1i and 10 nm for Fig. 2c-1ii). The ultraviolet–visible spectra of each solution show varying spectra (silver plasmon resonance peak is ∼400 nm) depending on the silver-shell thickness (Fig. 2b). As the silver shell gets thicker, the 400 nm peak becomes more dominant. The HRTEM images of the individual GSNDs shown in Fig. 2c-2, 2c-3 and 2c-4 strongly support the successful formation of corresponding structures. Distance measurement was based on HRTEM images (Fig. 2a, inset and Fig. 2c). The definite contrast difference in the HRTEM image enabled clear identification of the Au core and Ag shell32,37. The images show that, as the silver shell gets thicker, the particle size increases and the distance (dc−c) between the Au nanoparticle cores linearly increases (d1c−c (∼26 nm) < d2c−c (∼30 nm) < d3c−c (∼35 nm) < d4c−c(∼48 nm)). On the other hand, the interparticle gap distance (ds−s) between the Au nanoparticle surfaces decreases from ∼3.5 nm (d1c−c) in the Au nanoparticle dimer to ∼1.25 nm (d2c−c) in the GSND with a 3 nm shell thickness. In the case of >3 nm Ag shell thickness, no clear gap could be observed (d3s−s and d4s−s).

a, Ultraviolet–visible spectra before and after Au nanoparticle dimer formation and the corresponding TEM (upper) and HRTEM images (lower) of Au nanoparticle dimers. b, Ultraviolet–visible spectra while increasing the amount of AgNO3 solution added to the Au nanoparticle dimers. c, The typical HRTEM images of the synthesized nanoparticles, Au–Ag core–shell monomer with 5 nm (c-1i) and 10 nm Ag shell (c-1ii), and the GSND particles with ∼3 nm, ∼5 nm and ∼10 nm Ag shell thicknesses (C2, C3 and C4), respectively. The ds−s and dc−c indicate the distances between two particle surfaces and cores, respectively.
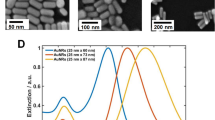
Next, we carried out AFM-correlated SERS measurements38,39 for the synthesized nanoparticles. First, an aliquot (20 μl) of nanoparticle solution was spin-coated on a poly-L-lysine-modified glass surface. The surface was then washed copiously with NANOpure water (18 MΩ cm−1), and the sample was spin-dried. The laser focal spot was exactly matched with the centre of the AFM tip for symmetrical scattering on the AFM tip end. The tapping-mode AFM module on top of the optical microscope stage was used to correlate the Raman signal with the AFM topographical image with an overlap precision of <100 nm. Figure 3a shows the magnified AFM images (1×1 μm2) of the core–shell monomer and heterodimer nanostructures. Figure 3b shows the correlated Raman spectra from the corresponding single AFM-imaged particles in Fig. 3a. No Raman signals were detected for the monomeric Au–Ag core–shell nanoparticles with 5 nm (Fig. 3a-1) and 10 nm (Fig. 3a-2) silver shells, respectively. The Au nanoparticle heterodimers (Fig. 3a-3; d1s−s, ∼3.5 nm) without a silver shell did not show any detectable Raman signal either. Raman signals from these structures were undetectable for both 514.5 nm and 632.8 nm laser sources. Owing to the steric repulsions of double-stranded DNA (30 base pairs) between the dimeric Au nanoparticles, close contact of these two nanoparticles is very unlikely, even in a dried state. Next, target-DNA-captured GSNDs were imaged and measured. When the Ag shell thickness was <3 nm (d2s−s=∼1.5 nm, Fig. 3a-4), no Raman signals were detected even after using an elevated incident laser power (∼200 μW). We suspect that a thin silver shell (<3 nm) could not make a proper junction for sufficient electromagnetic enhancement in SERS to generate a detectable Raman signal. However, when the Ag shell thickness was ∼5 nm (d3s−s∼0 nm), a Raman signal from a Cy3 label, located in the junction of two core–shell particles, was observed (Fig. 3a-5 and 3b-5). The characteristic Raman peaks for Cy3 dye were observed at 1,470 and 1,580 cm−1 from a 514.5 nm laser excitation40,41. The low intensity of these peaks is probably due to the presence of only one molecule within the hot junction region and relatively low laser power (100 μW) compared with the power intensity (in the milliwatt range) used in other single-molecule SERS studies7,8,9,10,11,12,13. It is known that the Raman intensity and spectral positions of Cy3 molecules fluctuate with time, and the Raman spectra are different for each observed nanostructure7,8,11,13,42. Therefore, spectral patterns are comparable but not fully compatible with the reported ones. We compared the Raman spectrum obtained from the GSND with 5 nm Ag shell thickness (Fig. 3b-5) with the Raman spectra of Cy3-modified oligonucleotides [5′-HS-(CH2)6-A10-PEG18-ATCCTTATCAATATTAAA-Cy3-3′, 1 nM, upper red line in Fig. 3c] and Cy3-free oligonucleotides [5′-HS-(CH2)6-A10-PEG18-ATCCTTATCAATATTAAA-3′, 1 μM, lower black line in Fig. 3c] in salt-induced Ag nanoparticle aggregates. The characteristic peaks for Cy3 dye appeared at 1,470 and 1,580 cm−1 (from Fig. 3b-5) and are in good agreement with those in Fig. 3c (upper red line in Fig. 3c), which is strong evidence for the existence of Cy3 molecules in the GSND40,41. The Raman spectrum of Cy3-free oligonucleotides (black line) features adenine peaks (734 and 1,320 cm−1; ref. 43). It was reported that the lowest detection limit for non-adenine DNA bases is in the sub-micromolar range44. The GSND with a 10 nm silver shell (Fig. 3a-6) shows dominant adenine peaks at 734 and 1,320 cm−1 along with a Cy3 peak at 1,480 cm−1. Importantly, some of these structures did not show any detectable Raman signals. We suspect that these results are due to a variety of reasons, including misplacement of the Cy3 molecule and improper and less reproducible junction structure with a thicker silver shell for the maximum SERS effect. Finally, our results suggest that the Cy3-modified DNA-tethered GSND with a 5 nm Ag shell (d3c−c∼35 nm, d3s−s∼0 nm) is a hot Raman structure with single-molecule sensitivity, and silver-shell-based close contact within the GSND junction was a critical factor in producing an effective electromagnetic enhancement in SERS. To prove our scheme works for other Raman dyes, we tethered GSNDs using carboxyfluorescein (FAM)- and 4,4-dimethylaminoazobenzene-4′-carboxylic acid (Dabcyl)-labelled DNA molecules, respectively, instead of Cy3-labelled DNA (note that FAM is a resonant Raman tag and Dabcyl is a non-resonant Raman tag). Both FAM- and Dabcyl-labelled GSNDs with a 5 nm Ag shell showed detectable Raman signals, and their spectra are well matched with reference spectra acquired from salt-induced Ag nanoparticle aggregates (Supplementary Fig. S5).

a, Atomic force micrograph (1×1 μm2) of the Au–Ag core–shell monomer, Au nanoparticle dimer and GSNDs to be measured. b, The corresponding SERS spectra taken from the nanostructures in a. All spectra were taken with a 514.5 nm excitation laser, 1 s accumulation, 100 μW at the sample and a 250 nm laser focal diameter. c, Raman spectra taken from Cy3-modified oligonucleotides (red line) and Cy3-free oligonucleotides (black line) in NaCl-aggregated silver colloids.
Next, to rigorously test the Raman data reproducibility of this hot Raman nanostructure, we put many GSNDs with 5-nm Ag shells on the same surface and analysed their Raman spectra (Fig. 4). Considering that the incident laser light is not exactly polarized to the interparticle axes of the dimer (panels 1–5 in Fig. 4a), relatively clear Raman signals from all five GSNDs on the same surface were detected. In the case of Fig. 4a-5 (the dimer orientation is nearly perpendicular to the incident light), a very small Raman peak at 1,470 cm−1 was detected. These results suggest that these individual DNA-tethered GSNDs generate Raman signals reproducibly and it is highly likely that single-molecule detection is possible repeatedly using this strategy.

a, The tapping-mode AFM images (5 μm×5 μm) of the GSNDs. The incident laser-light polarization direction is shown in the black circle. b, The SERS spectra from the corresponding GSNDs in a.
To prove that single-DNA detection is possible from a single GSND structure, several characterization experiments were carried out. Although many single-molecule verification experiments such as strong intensity fluctuation1,2,12,45,46,47, sudden replacement of analyte spectrum by a background spectrum8 in SERS, polarization properties of the SERS signal1,6, Poisson distribution of the SERS intensities, bi-analyte approach9,10 and frequency-domain approach12 have been developed, it is still controversial that these pieces of evidence could be considered as definite characteristics of single-molecule emission9,12,17,45,46,47. This uncertainty mainly arises from the difficulty of placing an analyte molecule at the rare hot Raman site and the lack of assurance that the signal comes only from this molecule. In this regard, our GSND could be an excellent model system for single-molecule detection using a single nanostructure. We tested the GSND with a 5 nm Ag shell with the following experimental procedures. First, a time-dependent reversible on/off blinking behaviour was observed in this structure with a reduced laser power (40 μW), and irreversible photobleaching had not occurred for a long-time signal observation. However, irreversible photobleaching was observed when higher laser power (>100 μW) was applied. An on–off cycle of Raman signals, indicative of the presence of a single molecule, was clearly observed as shown in Fig. 5a. Moreover, a brief on/off period appears suddenly and repeatedly (Fig. 5b). Furthermore, we observed the Raman intensity fluctuation on a second timescale owing to a single-molecule diffusion that makes this molecule Raman-active. The blinking phenomenon can be considered as a characteristic of single-molecule behaviour9,11,12, and we generally observed the random blinking behaviours in SERS spectra from the GSND (Fig. 5a,b). The GSND with a 5 nm Ag shell shows strong intensity fluctuation phenomena much like in other single-molecule experiments1,2,8,13,46. Next, we studied the polarization of the DNA-tethered GSND with a 5 nm Ag shell. Figure 5c,d shows the incident laser polarization dependence of the Raman signals for the single GSND. All of the spectra were taken with a 514.5 nm excitation laser, 20 s accumulation time and 40 μW laser power. Strong Cy3 peaks were observed when the incident laser light was polarized parallel to the longitudinal axis of the heterodimer, whereas negligible Raman signals were obtained otherwise (Fig. 5c). Polar plots for 1,470 and 1,580 cm−1 are shown in Fig. 5d. When the laser light was rotated by 20∘–90∘ away from the longitudinal axis, the Cy3 Raman signal was gradually reduced. The Raman peaks disappeared when the laser polarized perpendicular to the longitudinal axis (that is, 90∘ and 270∘). These polarization results are also in good agreement with previous reports providing evidence for single-molecule detection1,16,18,42.

a, Blinking SERS spectra taken from the GSND (accumulation time = 1 s, 100 times). b, The selected SERS spectra at 1,470 and 1,580 cm−1 from a. c, Polarization study results of the GSND with different incident-laser polarizations. d, Polar plots of integrated SERS intensities of the 1,470 and 1,580 cm−1 Raman bands with respect to θ (laser wavelength, 514.5 nm; laser power, ∼40 μW; laser focal diameter, ∼250 nm; accumulation time, 20 s at the sample).
Finally, the enhancement factor of the GSND with a 5 nm Ag shell was measured and calculated. As supported by a series of experimental results, it is likely that this single nanostructure generates a detectable Raman signal through the gap-engineered junction-based SERS effect. The calculated enhancement factor ( ) for the GSND was 2.7×1012, and details of its calculation are described in Supplementary Fig. S6. According to recent accurate experimental determinations of single-molecule enhancement factors using bi-analyte SERS and temperature-dependent SERS vibrational pumping methods17,47, an enhancement-factor value of the order of 107–108 is sufficient for the detection of a single molecule. It was also shown that maximum SERS enhancement factors are of the order of 1010 in typical SERS conditions and are at most of the order of 1012 in the best possible conditions17,47. Considering these reports and the absence of chemical enhancement in this measurement, we believe that our enhancement factor is large enough for single-molecule detection. Along with the precise gap-engineering of the GSND, the enormous electromagnetic enhancement of this nanostructure arises partly from the strong electronic effects of the core gold atoms in the core–shell structure, which could affect the surface silver atoms by a charge-transfer mechanism48.
) for the GSND was 2.7×1012, and details of its calculation are described in Supplementary Fig. S6. According to recent accurate experimental determinations of single-molecule enhancement factors using bi-analyte SERS and temperature-dependent SERS vibrational pumping methods17,47, an enhancement-factor value of the order of 107–108 is sufficient for the detection of a single molecule. It was also shown that maximum SERS enhancement factors are of the order of 1010 in typical SERS conditions and are at most of the order of 1012 in the best possible conditions17,47. Considering these reports and the absence of chemical enhancement in this measurement, we believe that our enhancement factor is large enough for single-molecule detection. Along with the precise gap-engineering of the GSND, the enormous electromagnetic enhancement of this nanostructure arises partly from the strong electronic effects of the core gold atoms in the core–shell structure, which could affect the surface silver atoms by a charge-transfer mechanism48.
Here, we showed that these Raman-active GSNDs have single-molecule sensitivity with high structural reproducibility. These results are important for the following reasons. First, our DNA-directed and magnetic separation-based nanostructure synthetic scheme opens opportunities in the high-yield synthesis of specific nanostructures for materials science and bio-detection applications (in this case, heterodimeric nanoparticles were synthesized with nearly 80% yield). Second, unlike the conventional strong electrolyte-induced nonspecific nanoparticle aggregation, our DNA-directed nanodimer assembly method can be easily scalable to produce targeted SERS-active nanoprobes. Third, we established a silver-shell coating-based nanogap-engineering method. Fourth, the nanogap-engineering of GSNDs allows for exploring hot SERS structures in an efficient and straightforward fashion. Fifth, our synthetic and detection strategies provide new ways of overcoming long-standing problems in Raman and materials-research societies about controlling the nanometre-gap, nanogeometry and dye position and environment with high reliability and reproducibility. This could eventually lead to large cross-section area-based Raman sensing for highly sensitive and quantitative Raman-based detection. Finally, these SERS-active GSNDs could be further modified by other biomolecules (such as proteins) and used as both in vitro and in vivo bio-labelling probes with ultrahigh sensitivity, quantification potential and multiplexing capability.
Methods
Synthesis of oligonucleotide-modified Au nanoparticles.
Freshly reduced thiolated oligonucleotides were used for Au nanoparticle probe preparation. A mixture of probe A protecting sequence [5′-CACGCGTTTCTCAAA-PEG18-A10-(CH2)3-SH-3′, 297 μl of 20.5 μM] and probe A target-capturing sequence [5′-TAACAATAATCCCTC-PEG18-A10-(CH2)3-SH-3′, 3.83 μl of 16.1 μM] were conjugated to 20-nm gold nanoparticles (2.0 ml of 1.03 nM). A mixture of probe B protecting sequence [5′-HS-(CH2)6-A10-PEG18-AAACTCTTTGCGCAC-3′, 717 μl of 14.9 μM solution] and probe B target-capturing sequence [5′-HS-(CH2)6-A10-PEG18-ATCCTTATCAATATTAAA-Cy3-3′, 13.59 μl of 3.96 μM] were conjugated to 30 nm gold nanoparticles (5.5 ml of 0.32 nM). The mixed solution was wrapped in foil and placed on an orbital shaker at room temperature overnight. The solution was then adjusted to obtain a final phosphate concentration of 10 mM (pH 7.4) with 0.1% (wt/vol) SDS. The adjusted solution was further incubated on an orbital shaker for 30 min and was brought to 0.3 M NaCl by the addition of six aliquots of 2 M NaCl solution. After a brief vortexing, the colloids were allowed to stand overnight at room temperature. The solution was centrifuged (10,000 rpm, 15 min), the supernatant was removed and the precipitate was redispersed in 0.3 M PBS solution (pH 7.4; this procedure was repeated twice). Finally, the solution was characterized using an ultraviolet–visible spectrophotometer.
Sequence information.
The protecting sequence for probe A is 3′-HS-(CH2)3-A10-PEG18-AAACTCTTTGCGCAC−5′, the target-capturing sequence for probe A is 3′-HS-(CH2)3-A10-PEG18-CTCCCTAATAACAAT-5′ and the sequence for magnetic probe A is 3′-NH2-A10-PEG18-ATTGTTATTAGGGAG-5′ (Tm=38 ∘C). The protecting sequence for probe B is 5′-HS-(CH2)6-A10-PEG18-AAACTCTTTGCGCAC-3′, the target-capture sequence for probe B is 5′-HS-(CH2)6-A10-PEG18-ATCCTTATCAATATTAAA-Cy3-3′ and the sequence for magnetic probe B is 5′-NH2-A10-PEG18-TTTAATATTGATAAGGAT-3′ (Tm=40 ∘C). The A10-PEG18 sequences are spacers. Finally, the target-DNA sequence is 5’-GAGGGATTATTGTTAAATATTGATAAGGAT-3’ (anthrax oligonucleotide).
Instrumentation.
Raman spectra were recorded using an AFM-correlated nano-Raman microscope equipped with an inverted optical microscope (Axiovert 200, Zeiss) and a piezoelectric x–y sample scanner (Physik Instrumente) manipulated by an independent homemade scanning controller. The 514.5 nm line of an argon ion laser (Melles Griot) was used as the excitation source coupled with a single-mode optical fibre. A dichroic mirror (520DCLP, Chroma Technology Corp.) directs the excitation laser beam from 50 nW to 1 mW into the oil-immersion microscope objective (×100, 1.3 numerical aperture, Zeiss), which focuses the beam to a diffraction-limited spot (∼250 nm) on the upper surface of the cover glass slip. The AFM (Bioscope, Digital Instruments, Veeco Metrology Group) with a Nanoscope IV controller was mounted on a micro-mechanical stage. The background Raman signals were collected on a liquid-nitrogen-cooled (−125 ∘C) CCD (charge-coupled device). The scattering spectra were recorded in the range of 500–2,000 cm−1, in one acquisition, 1 s accumulations and 100 μW at the sample. All of the data were baseline-corrected by subtracting the background signals from Si.
References
Nie, S. & Emory, S. R. Probing single molecules and single nanoparticles by surface-enhanced Raman scattering. Science 275, 1102–1106 (1997).
Kneipp, K. et al. Single molecule detection using surface-enhanced Raman scattering (SERS). Phys. Rev. Lett. 78, 1667–1670 (1997).
Hu, H. et al. Unified treatment of fluorescence and Raman scattering processes near metal surfaces. Phys. Rev. Lett. 93, 243002 (2004).
Hao, E. & Schatz, G. C. Electromagnetic fields around silver nanoparticles and dimers. J. Chem. Phys. 120, 357–366 (2004).
Michaels, A. M., Jiang, J. & Brus, L. Ag nanocrystal junctions as the site for surface-enhanced Raman scattering of single rhodamine 6G molecules. J. Phys. Chem. B 104, 11965–11971 (2000).
Shegai, T. O. & Haran, G. Probing the Raman scattering tensors of individual molecules. J. Phys. Chem. B 110, 2459–2461 (2006).
Andersen, P. C., Jacobson, M. L. & Rowlen, K. L. Flashy silver nanoparticles. J. Phys. Chem. B 108, 2148–2153 (2004).
Doering, W. E. & Nie, S. M. Single-molecule and single-nanoparticle SERS: Examining the roles of surface active sites and chemical enhancement. J. Phys. Chem. B 106, 311–317 (2002).
Le Ru, E. C., Meyer, M. & Etchegoin, P. G. Proof of single-molecule sensitivity in surface enhanced Raman scattering (SERS) by means of a two-analyte technique. J. Phys. Chem. B 110, 1944–1948 (2006).
Sawai, Y., Takimoto, B., Nabika, H., Ajito, K. & Murakoshi, K. Observation of a small number of molecules at a metal nanogap arrayed on a solid surface using surface-enhanced Raman scattering. J. Am. Chem. Soc. 129, 1658–1662 (2007).
Jiang, J., Bosnick, K., Maillard, M. & Brus, L. Single molecule Raman spectroscopy at the junctions of large Ag nanocrystals. J. Phys. Chem. B 107, 9964–9972 (2003).
Dieringer, J. A., Lettan, R. B. II, Scheidt, K. A. & Van Duyne, R. P. A frequency domain existence proof of single-molecule surface-enhanced Raman spectroscopy. J. Am. Chem. Soc. 129, 16249–16256 (2007).
Bosnick, K. A., Jiang, J. & Brus, L. E. Fluctuations and local symmetry in single-molecule rhodamine 6G Raman scattering on silver nanocrystal aggregates. J. Phys. Chem. B 106, 8096–8099 (2002).
Rodríguez-Lorenzo, L. et al. Zeptomol detection through controlled ultrasensitive surface-enhanced Raman scattering. J. Am. Chem. Soc. 131, 4616–4618 (2009).
Camden, J. P. et al. Probing the structure of single-molecule surface-enhanced Raman scattering hot spots. J. Am. Chem. Soc. 130, 12616–12617 (2008).
Li, W., Camargo, P. H. C., Lu, X. & Xia, Y. Dimers of silver nanospheres: Facile synthesis and their use as hot spots for surface-enhanced Raman scattering. Nano Lett. 9, 485–490 (2009).
Etchegoin, P. G. & Le Ru, E. C. A perspective on single molecule SERS: Current status and future challenges. Phys. Chem. Chem. Phys. 10, 6079–6089 (2008).
Qian, X.-M. & Nie, S. M. Single-molecule and single-nanoparticle SERS: From fundamental mechanisms to biomedical applications. Chem. Soc. Rev. 37, 912–920 (2008).
Fang, Y., Seong, N.-H. & Dlott, D. D. Measurement of the distribution of site enhancements in surface-enhanced Raman scattering. Science 321, 388–392 (2008).
Alivisatos, A. P. et al. Organization of ‘nanocrystal molecules’ using DNA. Nature 382, 609–611 (1996).
Zhang, J. P., Liu, Y., Ke, Y. G. & Yan, H. Periodic square-like gold nanoparticle arrays templated by self-assembled 2D DNA nanogrids on a surface. Nano Lett. 6, 248–251 (2006).
Deng, Z. X., Tian, Y., Lee, S. H., Ribbe, A. E. & Mao, C. D. DNA-encoded self-assembly of gold nanoparticles into one-dimensional arrays. Angew. Chem. Int. Ed. 44, 3582–3585 (2005).
Pinto, Y. Y. et al. Sequence-encoded self-assembly of multiple-nanocomponent arrays by 2D DNA scaffolding. Nano Lett. 5, 2399–2402 (2005).
Mirkin, C. A., Letsinger, R. L., Mucic, R. C. & Storhoff, J. J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 382, 607–609 (1996).
Nykypanchuk, D., Maye, M. M., van der Lelie, D. & Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nature 451, 549–552 (2008).
Park, S. Y. et al. DNA-programmable nanoparticle crystallization. Nature 451, 553–556 (2008).
Xiong, H. M., van der Lelie, D. & Gang, O. DNA linker-mediated crystallization of nanocolloids. J. Am. Chem. Soc. 130, 2442–2443 (2008).
Bidault, S., Javier Garcia de Abajo, F. & Polman, A. Plasmon-based nanolenses assembled on a well-defined DNA template. J. Am. Chem. Soc. 130, 2750–2751 (2008).
Zanchet, D. et al. Electrophoretic and structural studies of DNA-directed Au nanoparticle groupings. J. Phys. Chem. 106, 11758–11763 (2002).
Claridge, S. A., Liang, H. W., Basu, S. R., Frechet, J. M. & Alivisatos, A. P. Isolation of discrete nanoparticle-DNA conjugates for plasmonic applications. Nano Lett. 8, 1202–1206 (2008).
Maye, M. M., Nykypanchuk, D., Cuisnier, M., Lelie, D. & Gang, O. Stepwise surface encoding for high-throughput assembly of nanoclusters. Nature Mater. 8, 388–391 (2009).
Lim, D.-K., Kim, I.-J. & Nam, J.-M. DNA-embedded Au/Ag core–shell nanoparticles. Chem. Commun. 5312–5314 (2008).
Hurst, S. J., Lytton-Jean, A. K. R. & Mirkin, C. A. Maximizing DNA loading on a range of gold nanoparticle sizes. Anal. Chem. 78, 8313–8318 (2006).
Loweth, C. J., Caldwell, W. B., Peng, X., Alivisatos, A. P. & Schultz, P. G. DNA-based assembly of gold nanocrystals. Angew. Chem. 38, 1808–1812 (1999).
Moreau, J. W. & Sharp, T. G. A transmission electron microscopy study of silica and kerogen biosignatures in-1.9 Ga gunflint microfossils. Astrobiology 4, 196–210 (2004).
Park, S.-J., Lazarides, A. A., Storhoff, J. J., Pesce, L. & Mirkin, C. A. The structural characterization of oligonucleotide-modified gold nanoparticle networks formed by DNA hybridization. J. Phys. Chem. B 108, 12375–12380 (2004).
Liu, M. & Guyot-Sionnest, P. Synthesis and optical characterization of Au/Ag core/shell nanorods. J. Phys. Chem. B 108, 5882–5888 (2004).
Suh, Y. D., Schenter, G. K., Zhu, L. & Lu, H. P. Probing nanoscale surface enhanced Raman-scattering fluctuation dynamics using correlated AFM and confocal ultramicroscopy. Ultramicroscopy 97, 89–102 (2003).
Lee, Y. M., Jeon, K.-S., Suh, Y. D. & Choi, H. C. Silencing of metallic single-walled carbon nanotubes via spontaneous hydrosilylation. Small 5, 1398–1402 (2009).
Faulds, K., Smith, W. E. & Graham, D. Evaluation of surface-enhanced resonance Raman scattering for quantitative DNA analysis. Anal. Chem. 76, 412–417 (2004).
Stokes, R. J. et al. Quantitative enhanced Raman scattering of labeled DNA from gold and silver nanoparticles. Small. 3, 1593–1601 (2007).
Imura, K., Okamoto, H., Hossain, M. K. & Kitajima, M. Visualization of localized intense optical fields in single gold-nanoparticle assemblies and ultrasensitive Raman active sites. Nano Lett. 6, 2173–2176 (2006).
Barhoumi, A., Zhang, D., Tam, F. & Halas, N. J. Surface-enhanced Raman spectroscopy of DNA. J. Am. Chem. Soc. 130, 5523–5525 (2008).
Bell, S. E. J. & Sirimuthu, N. M. S. Surface-enhanced Raman spectroscopy (SERS) for sub-micromolar detection of DNA/RNA mononucleotides. J. Am. Chem. Soc. 128, 15580–15581 (2006).
Moskovits, M., Tay, L.-L., Yang, J. & Haslett, T. SERS and the single molecule. Top. Appl. Phys. 82, 215–226 (2002).
Weiss, A. & Haran, G. Time-dependent single-molecule Raman scattering as a probe of surface dynamics. J. Phys. Chem. B 105, 12348–12354 (2001).
Le Ru, E. C., Blakie, E., Meyer, M. & Etchegoin, P. G. Surface enhanced Raman scattering (SERS) enhancement factors; a comprehensive study. J. Phys. Chem. C 111, 13794–13803 (2007).
Yang, Y., Shi, J., Kawamura, G. & Nogami, M. Preparation of Au–Ag, Ag–Au core–shell bimetallic nanoparticles for surface-enhanced Raman scattering. Scr. Mater. 58, 862–865 (2008).
Acknowledgements
J.-M.N. was supported by the 21C Frontier Functional Proteomics Project (FPR08-A2-150), the Nano R&D programme (2008-02890) and the Basic Science Research Program (2009-0077361) through the National Research Foundation of Korea (NRF) from the Ministry of Education, Science and Technology. Y.D.S. was supported by KRICT (KK-0904-02), the Nano R&D Program (No. 2009-0082861), the Pioneer Research Center Program of NRF (No. 2009-0081511), the Development of Advanced Scientific Analysis Instrumentation Project of KRISS by MEST and the Eco-technopia 21 Project by KME. We would also like to acknowledge the Industrial Core Technology Development Program by the Ministry of Knowledge Economy (No. 10033183) for financial support.
Author information
Authors and Affiliations
Contributions
Y.D.S. and J.-M.N. conceived the initial idea. J.-M.N. designed synthetic schemes for nanoprobes and D.-K.L. and J.-M.N. synthesized and characterized nanoprobes. Raman spectra and AFM images were obtained by K.-S.J. and D.-K.L. under the supervision of Y.D.S. and J.-M.N. Single-molecule experiments were designed and guided by Y.D.S. and J.-M.N., and carried out by K.-S.J. and D.-K.L. H.M.K. measured and calculated the enhancement factor. J.-M.N., D.-K.L. and Y.D.S. wrote the article with partial contribution from K.-S.J. and H.M.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Information (PDF 659 kb)
Rights and permissions
About this article
Cite this article
Lim, DK., Jeon, KS., Kim, H. et al. Nanogap-engineerable Raman-active nanodumbbells for single-molecule detection. Nature Mater 9, 60–67 (2010). https://doi.org/10.1038/nmat2596
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmat2596
This article is cited by
-
Dynamical control of nanoscale light-matter interactions in low-dimensional quantum materials
Light: Science & Applications (2024)
-
Efficient optical plasmonic tweezer-controlled single-molecule SERS characterization of pH-dependent amylin species in aqueous milieus
Nature Communications (2023)
-
Cucurbit[8]uril-mediated SERS plasmonic nanostructures with sub-nanometer gap for the identification and determination of estrogens
Microchimica Acta (2023)
-
Highly sensitive near-infrared SERS nanoprobes for in vivo imaging using gold-assembled silica nanoparticles with controllable nanogaps
Journal of Nanobiotechnology (2022)
-
Three-dimensional nanoframes with dual rims as nanoprobes for biosensing
Nature Communications (2022)
