
Abstract
The heterogeneity of HIV and the different human leukocyte antigen (HLA) backgrounds of infected individuals have posed challenges to understanding the pathogenesis of HIV infection. But continuing advances in our knowledge of the role of immune responses in controlling HIV viremia should help to define goals for immune-based therapies and vaccine strategies against AIDS.
Similar content being viewed by others
Main
AIDS is essentially an infection of the immune system. The first reported cases of this syndrome1 were seen in young adults afflicted with opportunistic infections that, until then, had only been seen in the setting of profound immune deficiency. This was followed by the rapid identification of HIV-1 as the causative agent of AIDS2, the development of a nonhuman primate model of AIDS virus infection3 and the detection of immune responses to HIV-1 in infected persons4,5,6. Since then, global research efforts have led to detailed characterization not only of the effects of the virus on the host, but also an understanding of the ultimate failure of the immune system to contain the infection. The complex interactions between virus and immune system have been unraveled through experimental models of AIDS virus infection in nonhuman primates, as well as studies of infected humans. Indeed, with regards to breadth and specificity of immune responses, it is likely that more information has been generated concerning HIV and simian immunodeficiency virus (SIV) than any other viruses in history. This review, which by no means is able to reference all of the important contributions over the past 20 years, highlights our present understanding of AIDS immunopathogenesis, emerging advances in immunotherapy and remaining key research questions for the future.
Antibody responses to HIV-1
Although antibody responses have a central role in clearing many viral infections, accumulating data suggest that this may not be true for HIV-1 infection. Antibodies in the sera of HIV-1-infected individuals have only weak neutralizing activity for primary HIV-1 isolates7,8,9, with most of the antibodies being non-neutralizing and directed at virion debris10. In addition, the burst of HIV-1 replication that occurs in the first days after initial infection is contained in the infected individual well before the development of an antibody that can neutralize the virus11. In fact, depletion of B lymphocytes in rhesus monkeys by infusion of monoclonal antibodies to CD20 significantly delayed the emergence of a virus-neutralizing antibody response after SIV infection, but did not alter the kinetics of early viral clearance12. These observations suggest that neutralizing antibody may not be important in the early control of HIV-1 replication.
Antibodies that neutralize HIV-1 recognize one of three distinct neutralizing domains of the HIV-1 envelope: the third hypervariable (V3) loop of the envelope glycoproteins, the CD4 binding sites of the envelope and the transmembrane gp41 protein. Given the importance of the V3 loop in the interactions of the HIV-1 envelope with chemokine receptors, it is not surprising that antibodies that bind to this domain of envelope can inhibit viral infection of cells13. Antibodies specific for the V3 loop are the first neutralizing antibodies that arise in HIV-1-infected individuals14. However, this domain is problematic as a target for vaccine-elicited, broadly neutralizing antibodies because V3 loop–specific antibodies are, in general, isolate-specific in their neutralizing ability. Moreover, extensive glycosylation of the envelope on primary HIV-1 isolates is likely to render them poorly accessible to antibodies15. The CD4 binding domains of the HIV-1 envelope are highly conserved among viral isolates, and antibodies that bind to these domains are therefore reactive with a diversity of viruses. However, antibodies specific for the CD4 binding site are only weakly neutralizing. The sequence of the transmembrane gp41 protein is highly conserved among HIV-1 isolates, and monoclonal antibodies to gp41 that neutralize a variety of HIV-1 isolates have been described16. This region of the virus may therefore be a good target for vaccine-induced neutralizing antibody responses.
A number of studies suggest that neutralizing antibodies contribute little to the control of HIV-1 replication in individuals with established infections, despite the finding that they exert immune selection pressure17,18. Immunodeficient mice reconstituted with human lymphoid tissue have been infected with HIV-1 and then evaluated after infusion with neutralizing HIV-1-specific monoclonal antibodies. These antibody treatments had little effect on viral replication in this model10. Similarly, in HIV-1-infected individuals, intravenous infusion of hyperimmune globulin with high titers of HIV-1-specific antibodies had little effect on viral load or disease progression19. However, pre-exisiting circulating neutralizing antibody has been shown to alter the clinical outcome of SIV and HIV/SIV hybrid (SHIV) infections in macaques. Infusion of either serum IgG or combinations of monoclonal antibodies that neutralize these viruses attenuates the pathogenicity or even blocks the establishment of infection by these lentiviruses20,21,22. The fact that neutralizing antibodies seem to be able to entirely protect against initial infection suggests that such antibodies will be very important in any strategy to prevent HIV-1 infection.
Cellular immune responses to HIV-1
In contrast to the above observations concerning neutralizing antibodies and virus containment, virus-specific CD8+ cytotoxic T lymphocytes (CTLs) have been implicated in the control of HIV-1 replication. HIV-1-specific CTLs have been found in large numbers in a variety of anatomic compartments in both HIV-1-infected humans and SIV-infected macaques, including peripheral blood, bronchoalveolar spaces, lymph nodes, spleen, skin, cerebrospinal fluid, semen and both vaginal and gastrointestinal mucosal tissue. Moreover, CD8+ T lymphocytes can inhibit HIV-1 replication in vitro23. Multiple mechanisms have been associated with this antiviral effect. CTL can lyse HIV-1-infected cells in vitro and block propagation of the infection24. These effector cells also produce soluble factors that can mediate this effect23,25. The β-chemokines MIP-1α, MIP-1β and RANTES have activity against HIV-1 (ref. 26), colocalize with granzymes and perforin, and are coordinately secreted by CTLs after being triggering by antigen encounter27. Other soluble factors may also have a role in this cell-mediated inhibition of viral replication28.
Partial control of viral replication occurs during the early days after infection and correlates temporally with the emergence of an HIV-1-specific CD8+ CTL response. An association was shown between the appearance of effector cell populations that lyse HIV-1-expressing target cells and the decline in plasma viral RNA during the period of primary HIV-1 infection29,30,31,32. Consistent with this observation, oligoclonal populations of T lymphocytes expand markedly in the peripheral blood of infected individuals at the time of virus containment in the early weeks after HIV-1 infection33. These populations of T lymphocytes are likely to represent clonally restricted CTLs. Studies have also been done using assays to evaluate populations of CD8+ T cells for killing, clonality and tetramer binding in SIV-infected macaque models; these studies show a clear temporal association between the expansion of CTLs and the clearance of virus34,35,36. More detailed studies, however, have been largely unable to show a clear association between viral load and breadth or magnitude of CTL responses in humans37,38,39, perhaps because of inaccurate measurements made using reference strains of virus rather than autologous virus40.
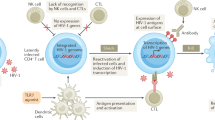
Perhaps the most compelling evidence for the importance of CD8+ CTLs in containing HIV-1 replication comes from studies in SIV-infected rhesus monkeys. In vivo depletion of CD8+ cells in monkeys, achieved by infusion of monoclonal antibodies to CD8, had profound effects on the replication of SIV41,42. When the duration of depletion was greater than 28 days, primary viremia was never cleared after infection and the monkeys died with a rapidly progressive AIDS-like syndrome (Fig. 1). In addition, transient CD8+ lymphocyte depletion of chronically SIV-infected rhesus monkeys was associated with a substantial rise in viral replication that returned to baseline levels coincident with the re-emergence of the CD8+ cell population.

Katie Ris
Acute SIV infection in a monkey model of AIDS results in a peak of viremia that is normally partially contained by CTLs. When CTLs are depleted, viremia is not contained.
Nonhuman primate studies have also shown the ramifications of potent virus-specific CTL responses on the clinical course of AIDS. Several groups have recently shown that rhesus monkeys that were vaccinated to elicit CTL responses and then infected with SIV or SHIV had a more benign clinical course than unvaccinated monkeys43,44,45,46. These monkeys had lower viral loads, better-preserved populations of CD4+ T-lymphocytes and survived for longer than unvaccinated monkeys. In fact, the extent of clinical protection in the monkeys correlated with the magnitude of the vaccine-elicited CTL responses before infection. Thus, robust CTL responses confer significant protection against SIV and SHIV replication in monkeys.
Consistent with the importance of CTLs in controlling HIV-1, SIV and SHIV replication, the major histocompatibility complex (MHC) class I haplotypes of infected individuals has a significant predictive value for the rate of clinical disease progression. Because MHC class I molecules bind fragments of viral proteins and present those fragments to immune cells to initiate immune responses, the particular fragment of a virus that is immunogenic for CTLs and the magnitude of virus-specific CTL responses are determined in part by the MHC class I molecules expressed in an individual. For example, the SLYNTVATL fragment of HIV-1 Gag binds to the HLA-A2 molecule which efficiently presents it to immune cells, resulting in a relatively reproducible, high-frequency, Gag-specific CTL response in HLA-A2-positive individuals. Heterozygosity at class I alleles, as well as the expression of the MHC class I molecules HLA-B27 and HLA-B57, in infected individuals are associated with better clinical outcomes after HIV-1 infection47,48,49, whereas expression of a particular haplotype of HLA-B35 is associated with worse outcome50. Specific HLA alleles have now also been associated with vaccine responsiveness in HIV vaccine trials51. Similarly, rhesus monkeys that express the MHC class I molecule Mamu-A*01 have a more benign disease course after infection with some SIV and SHIV isolates than do other rhesus monkeys52. These observations underscore the importance of CTLs in containing HIV-1 replication and highlight the genetic constraints on immune control, the mechanism of which remains poorly understood.
Virus-specific CD4+ T lymphocytes also have an important role in controlling HIV-1 replication. Although assays to measure T-lymphocyte proliferation in response to viral antigen have shown little functional virus-specific CD4+ T-lymphocyte activity in HIV-1-infected individuals, more sensitive assays for measuring cytokine production by viral peptide–stimulated lymphocytes have shown that many HIV-1-infected individuals do indeed have virus-specific CD4+ T-lymphocyte populations53,54. Studies in a nonhuman primate model have shown that oligoclonal populations of CD4+ T lymphocytes can be detected in vivo for prolonged periods of time in chronically infected monkeys, a finding consistent with the persistence of viral epitope–specific CD4+ T lymphocytes55. Moreover, the magnitude of CD4+ T-lymphocyte proliferation and cytokine production correlate with the clinical status of HIV-1-infected humans and SIV- or SHIV-infected monkeys56,57. Because there is little evidence that CD4+ T lymphocytes have a role as effector cells in this setting, these cells are helping to facilitate CTL and antibody responses.
Immune escape
A central unanswered question is why replication of the AIDS virus, despite the induction of cellular and humoral immune responses after infection, is not contained and leads to progressive and ultimately profound immune suppression. Although numerous reasons for lack of immune control have been proposed, the best documented has been immune escape through the generation of mutations in targeted epitopes of the virus. When effective selection pressure is applied, the error-prone reverse transcriptase and high replication rate of HIV-1 allow for rapid replacement of circulating virus by those carrying resistance mutations as was first observed with administration of potent antiretroviral therapy.
Selection pressure exerted by humoral and cellular immune responses to HIV-1 is well documented, but its precise contribution to immune failure is still not clear. Selection pressure by neutralizing antibodies can be observed in vitro58 and is apparent in vivo early in infection, as shown by the emergence of virus that is able to evade early autologous neutralizing antibodies even though it remains sensitive to neutralization by control sera59 (Fig. 2). Studies using recombinant virus assays have shown that the rate of neutralizing-antibody escape exceeds the rapid rate of change observed with drug selection pressure, and can account for the extensive variability in the envelope protein compared with other genes17,18. The mechanism of escape may involve changes in envelope glycans that shield antibody binding sites by steric hindrance18. These studies clearly show that neutralizing antibodies exert considerable selection pressure, and that fully functional envelope variants that escape immune detection continuously emerge and become the dominant circulating species. Despite the clear induction of antibody escape, however, a direct link between the degree of antibody escape and disease progression remains to be shown.

Katie Ris
After acute infection, virus-specific neutralizing antibodies are slow to develop and type-specific, and exert selection pressure. The virus rapidly escapes by generating new variants that are not recognized by the initial antibodies. As antibodies to the emerging variants develop, the virus mutates further and thus continues to evade neutralizing antibodies.
Viral escape from CTL responses is another mechanism of immune escape that has been documented during both acute31,32,60 and chronic61,62 infection. Escape occurs even through single amino-acid mutations in an epitope, at sites essential for MHC binding or T-cell-receptor recognition, but may also be influenced by mutations in flanking regions that affect antigen processing. The potentially strong immune selection pressure exerted by CTLs has been particularly well demonstrated in acute SIV infection, in which SIV-infected monkeys generated strong initial CTL responses against an epitope in Tat60. Although the infecting virus was apparently controlled by an effective CTL response against an early-expressed Tat epitope, new viruses with mutations in Tat emerged as this Tat-specific CTL response was being generated, and the variant viruses went on to establish chronic uncontrolled infection.
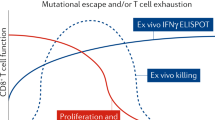
CTL escape has also been documented in transmission studies and after immunization and subsequent infection. Mothers who express HLA-B27, associated with long-term, nonprogressing HIV infection, transmitted a CTL escape variant to their children such that the epitope that is normally associated with protection in adults could not be targeted63. In contrast, children who inherited HLA-B27 from their fathers and HIV from their mothers received a virus that had not been under B27-restricted selection pressure and were able to mount vigorous CTL responses and achieve relative control of infection. Recent studies in macaques immunized with SHIV provide the most direct link between immune escape from CTLs and disease progression64. Immunized animals were not protected from infection but seemed to be protected from disease progression, in that viral load was contained in the setting of induction of potent SHIV-specific CTL responses. During prolonged follow-up, one animal developed an increasing viral load that which was temporally related to the emergence of a CTL escape mutation within a dominant epitope (Fig. 3). However, not all CTL responses seem to exert such pronounced selection pressure on the AIDS virus65.

Katie Ris
Shown are the rise in plasma viral RNA and fall in peripheral blood CD4+ T-lymphocyte count in the animal. The mutation that appeared in the targeted epitope at week 20 was not recognized by the CTL response.
Evidence supporting the influence of CTL selection pressure on this virus also comes from population studies examining associations between HLA alleles and specific mutations. HLA-associated selection of mutations was found to be predictive of viral load when HIV reverse transcriptase sequences were examined in a cohort of over 400 individuals with chronic HIV-1 infection66. This evidence of HLA imprinting on a population level supports a significant role of CTL responses in driving HIV evolution67. The apparent advantage of rare HLA alleles is consistent with these findings—those individuals expressing rare alleles would be less likely to encounter viruses that had already developed fixed mutations in the dominant epitopes presented by that allele68. The finding that some alleles preferentially present epitopes to the immune system early in infection69, whereas others may not present until later in infection70, suggests that not all MHC alleles contribute equally to immune control and underscores our lack of understanding of the parameters that influence immunodominance.
Immune dysfunction
The finding that not all viral CTL epitopes develop escape mutations65,71,72 suggests that functional impairment of cellular immune responses may actually limit the selection pressure applied by this arm of the immune system73. There have been numerous proposed mechanisms for this immune dysfunction, but based on prior animal studies of immune failure in chronic viral infections, it is likely that lack of sufficient HIV-specific CD4+ T-helper cell proliferation and expansion is a crucial feature of this impairment53,54. In macaque models, there is a clear loss of the capacity to express cytokines, beginning as early as the time of peak viremia in acute infection57. The selective infection of HIV-1-specific CD4+ T cells in infected individuals provides a mechanistic explanation for loss of these cells early in infection74 and explains why these responses are restored with early treatment of acute infection56,75,76.
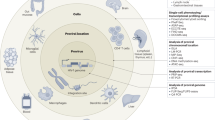
Other mechanisms of immune evasion have been noted, but the relative importance of their contribution to overall immune impairment is less certain (Fig. 4). Downregulation of HLA class I by Nef impairs CTL recognition77 and limits the inhibitory effects of CTLs on viral replication78. This effect is limited to HLA-A and HLA-B, which tend to be the dominant restricting alleles79. Defects in differentiation and maturation of CTLs80,81,82 may result in impaired in vivo function and may relate to a lack of CD4+ T-cell help. Other studies have shown that CTLs against HIV-1 are deficient in perforin or cytokine production81,83,84, but whether this is the result of an intrinsic defect or a recent encounter with antigen is not entirely clear. Another suggestion of immune impairment is the downmodulation of key signaling molecules for T-cell activation and costimulation85. More recent studies have shown that CD8+ T cells from infected individuals are able to secrete interferon-γ, but in those who do not control viremia, there is a defect in the ability of CD8+ T cells to proliferate in response to antigenic challenge81. Differences in viral replicative fitness may also affect the ability of the immune system to contain the virus. Dissecting the relative contributions of each of these potential mechanisms of immune impairment remains a challenge.

Katie Ris
Potential mechanisms of immune failure in HIV infection.
Immmunotherapy
The rationale for immune-based therapy in HIV-1 infection stems from the observation that prolonged highly active antiretroviral therapy (HAART) leads to increases in naive cells86, as well as from the improvement in observed functional defects in CD4+ and CD8+ T cells that are characteristic of this infection. The restoration of immune responsiveness to other pathogens such as cytomegalovirus with administration of HAART indicates that immune suppression is reversible after prolonged HIV infection. In contrast, despite small increases in viremia typically observed in individuals successfully treated with HAART, HIV-1-specific immunity is not enhanced but instead declines87,88. This suggests that the antigenic threshold required for induction of responses is not being achieved, and that the defect may be in the induction phase of the response.
Numerous approaches to addressing immune augmentation in HIV-1 infection are currently under way, but proof of principle that a clinical benefit can be achieved is lacking. Enough data to warrant discussion have been accrued by at least four approaches: adoptive therapy, cytokine therapy, therapeutic immunization and a combination of HAART and treatment interruption to boost immune responses to autologous virus. The short-term outcomes of each of these approaches will help to shape the future direction of the field.
Adoptive therapy has been done using both antibodies and cells. Infusion of cocktails of neutralizing monoclonal antibodies has led to marked protection from infection in nonhuman primates89. Infused antigen-specific CTLs can home to sites of virus replication90, and escape mutants are rapidly selected after adoptive transfer of Nef-specific CTL clones; these observations provide evidence of in vivo function of CTLs91. Infusion of interleukin-2 by a number of dosing schedules and routes has resulted in clear increases in CD4+ T-cell counts92, but after years of research it is still not clear whether this increase actually affects disease progression.
Immune augmentation has also been approached through antigen-specific enhancement by therapeutic immunization. Augmentation of CD8+ T-cell responses has been disappointing, with no consistent demonstration of immunogenicity and no clear impact on viral load93. Augmentation of virus-specific CD4+ T-cell responses has been achieved in studies of chronically infected individuals after increases in their naive cells through prolonged HAART therapy, but effects on immune control and viral load were lacking94. Arguably, the most notable report of immune augmentation to date involves the adoptive transfer of autologous dendritic cells pulsed with inactivated SIV95. After receiving injections of these dendritic cells, the chinese macaques experienced increases in virus-specific cellular immune responses and a more than 100-fold decrease in steady-state viremia. These findings, if confirmed, suggest that the defect in immune control may relate to the induction phase of the immune response, and would be consistent with recent studies of immune induction in the absence of CD4+ T-cell help96,97.
At least transient control of viremia has been achieved with early treatment of acute HIV-1 or SIV infection, followed by supervised periods of treatment interruption that have been associated with broadening and increased magnitude of cellular immune responses to the virus98,99. The same approach has been less successful in the setting of chronic HIV-1 infection100,101,102, probably because of increased virus variability and a greater chance for immune escape, as well as lack of restoration of virus-specific T-helper cell responses with HAART alone103. Late therapeutic failure has been observed, in at least one case, as a result of superinfection104. As yet, no studies have shown a clinical benefit to this approach.
Conclusions
The crucial roles of cellular and humoral immune responses in controlling HIV-1 viremia and influencing the viral set point are being elucidated, providing targets for immunotherapeutic intervention and defining goals for vaccine strategies. The true correlates of immune protection and immune failure need to be better defined—a task that is no doubt made more difficult by viral heterogeneity and the diverse HLA backgrounds of infected individuals which may influence the course of infection. Experiments in animal models of chronic viral infection help put this into perspective. In the lymphocytic choriomeningitis virus model, an inbred strain of mouse can entirely resolve infection when infected with the Armstrong stain of virus, whereas when the same strain is infected with the related Clone-13 virus, chronic infection persists for the duration of the animal's life. The only differences between these two viruses are three nucleotides and two amino acids105,106. Understanding HIV pathogenesis in the setting of tremendous viral and HLA diversity will be a challenge. Nevertheless, recent advances showing the ability of the immune system to at least partially contain HIV and SIV provide hope that research on AIDS vaccines and immune-based therapies may indeed bear fruit.
References
Friedman-Kien, A. et al. Kaposi's sarcoma and Pneumocystis pneumonia among homsexual men- New York City and California. MMWR 30, 305–308 (1981).
Barré-Sinoussi, F. et al. Isolation of a T-lymphotrophic retrovirus from a patient at risk for acquired immunodeficiency syndrome (AIDS). Science 220, 868–871 (1983).
Letvin, N.L. et al. Induction of AIDS-like disease in macaque monkeys with T-cell tropic retrovirus STLV-III. Science 230, 71–73 (1985).
Weiss, R.A. & Clapham, P.R. Neutralization of human T-lymphotropic virus type III by sera of AIDS and AIDS-risk patients. Nature 316, 69–72 (1985).
Plata, F. et al. AIDS virus-specific cytotoxic T lymphocytes in lung disorders. Nature 328, 348–351 (1987).
Walker, B.D. et al. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature 328, 345–348 (1987).
Montefiori, D.C. et al. Neutralizing and infection-enhancing antibody responses to human immunodeficiency virus type 1 in long-term nonprogressors. J. Infect. Dis. 173, 60–67 (1996).
Moog, C., Fleury, H.J., Pellegrin, I., Kirn, A. & Aubertin, A.M. Autologous and heterologous neutralizing antibody responses following initial seroconversion in human immunodeficiency virus type 1-infected individuals. J. Virol. 71, 3734–3741 (1997).
Moore, J.P. et al. Primary isolates of human immunodeficiency virus type 1 are relatively resistant to neutralization by monoclonal antibodies to gp120, and their neutralization is not predicted by studies with monomeric gp120. J. Virol. 69, 101–109 (1995).
Poignard, P. et al. Neutralizing antibodies have limited effects on the control of established HIV-1 infection in vivo. Immunity 10, 431–438 (1999).
Pilgrim, A.K. et al. Neutralizing antibody responses to human immunodeficiency virus type 1 in primary infection and long-term-nonprogressive infection. J. Infect. Dis. 176, 924–932 (1997).
Schmitz, J.E. et al. Effect of humoral immune responses on controlling viremia during primary infection of rhesus monkeys with simian immunodeficiency virus. J. Virol. 77, 2165–2173 (2003).
Choe, H. et al. The β-chemokines receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85, 1135–1148 (1996).
Javaherian, K. et al. Principal neutralizing domain of the human immunodeficiency virus type 1 envelope protein. Proc. Natl. Acad. Sci. USA 86, 6768–6772 (1989).
Reitter, J.N., Means, R.E. & Desrosiers, R.C. A role for carbohydrates in immune evasion in AIDS. Nat. Med. 4, 679–684 (1998).
Trkola, A. et al. Cross-clade neutralization of primary isolates of human immunodeficiency virus type 1 by human monoclonal antibodies and tetrameric CD4-IgG. J. Virol. 69, 6609–6617 (1995).
Richman, D.D., Wrin, T., Little, S.J. & Petropoulos, C.J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 100, 4144–4149 (2003).
Wei, X. et al. Antibody neutralization and escape by HIV-1. Nature 422, 307–312 (2003).
Jacobson, J.M. Passive immunization for the treatment of HIV infection. Mt. Sinai J. Med. 65, 22–26 (1998).
Mascola, J.R. et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat. Med. 6, 207–210 (2000).
Haigwood, N.L. et al. Passive immune globulin therapy in the SIV/macaque model: early intervention can alter disease profile. Immunol. Lett. 51, 107–114 (1996).
Baba, T.W. et al. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian–human immunodeficiency virus infection. Nat. Med. 6, 200–206 (2000).
Walker, C.M., Moody, D.J., Stites, D.P. & Levy, J.A. CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science 234, 1563–1566 (1986).
Tsubota, H., Lord, C.I., Watkins, D.I., Morimoto, C. & Letvin, N.L. A cytotoxic T lymphocyte inhibits acquired immunodeficiency syndrome virus replication in peripheral blood lymphocytes. J. Exp. Med. 169, 1421–1434 (1989).
Yang, O.O. et al. Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: evidence for HLA class I-restricted triggering of cytolytic and noncytolytic mechanisms. J. Virol. 71, 3120–3128 (1997).
Cocchi, F. et al. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 270, 1811–1815 (1995).
Wagner, L. et al. Beta-chemokines are released from HIV-1-specific cytolytic T-cell granules complexed to proteoglycans. Nature 391, 908–911 (1998).
Zhang, L. et al. Contribution of human alpha-defensin 1, 2, and 3 to the anti-HIV-1 activity of CD8 antiviral factor. Science 298, 995–1000 (2002).
Koup, R.A. et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68, 4650–4655 (1994).
Borrow, P., Lewicki, H., Hahn, B.H., Shaw, G.M. & Oldstone, M.B.A. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68, 6103–6110 (1994).
Borrow, P. et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3 (1997).
Price, D.A. et al. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 94, 1890–1895 (1997).
Pantaleo, G. et al. Major expansion of CD8+ T cells with a predominant V beta usage during the primary immune response to HIV. Nature 370, 463–467 (1994).
Yasutomi, Y., Reimann, K.A., Lord, C.I., Miller, M.D. & Letvin, N.L. Simian immunodeficiency virus-specific CD8+ lymphocyte response in acutely infected rhesus monkeys. J. Virol. 67, 1707–1711 (1993).
Chen, Z.W. et al. T cell receptor Vβ repertoire in an acute infection of rhesus monkeys with simian immunodeficiency viruses and a chimeric simian-human immunodeficiency virus. J. Exp. Med. 182, 21–31 (1995).
Kuroda, M.J. et al. Emergence of CTL coincides with clearance of virus during primary simian immunodeficiency virus infection in rhesus monkeys. J. Immunol. 162, 5127–5133 (1999).
Ogg, G.S. et al. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 279, 2103–2106 (1998).
Betts, M.R. et al. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: relationship to viral load in untreated HIV infection. J. Virol. 75, 11983–11991 (2001).
Addo, M.M. et al. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J. Virol. 77, 2081–2092 (2003).
Altfeld, M. et al. Enhanced detection of HIV-1-specific T cell responses to highly variable regions using peptides based on autologous virus sequences. J. Virol. (in the press).
Schmitz, J.E. et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283, 857–860 (1999).
Jin, X. et al. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189, 991–998 (1999).
Seth, A. et al. Immunization with a modified vaccinia virus expressing simian immunodeficiency virus (SIV) Gag-Pol primes for an anamnestic Gag-specific cytotoxic T-lymphocyte response and is associated with reduction of viremia after SIV challenge. J. Virol. 74, 2502–2509 (2000).
Amara, R.R. et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292, 69–74 (2001).
Barouch, D.H. et al. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 290, 486–492 (2000).
Shiver, J.W. et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 415, 331–335 (2002).
Kaslow, R.A. et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2, 405–411 (1996).
Migueles, S.A. et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. USA 97, 2709–2714 (2000).
Carrington, M. et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283, 1748–1752 (1999).
Gao, X. et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N. Engl. J. Med. 344, 1668–1675 (2001).
Kaslow, R.A. et al. Polymorphisms in HLA class I genes associated with both favorable prognosis of human immunodeficiency virus (HIV) type 1 infection and positive cytotoxic T-lymphocyte responses to ALVAC-HIV recombinant canarypox vaccines. J. Virol. 75, 8681–8689 (2001).
Pal, R. et al. ALVAC-SIV-gag-pol-env–based vaccination and macaque major histocompatibility complex class I (A*01) delay simian immunodeficiency virus SIVmac-induced immunodeficiency. J. Virol. 76, 292–302 (2002).
Pitcher, C.J. et al. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nat. Med. 5, 518–525 (1999).
McNeil, A.C. et al. High-level HIV-1 viremia suppresses viral antigen-specific CD4(+) T cell proliferation. Proc. Natl. Acad. Sci. USA 98, 13878–13883 (2001).
Chen, Z.W. et al. Prolonged dominance of clonally restricted CD4+ T cells in macaques infected with simian immunodeficiency viruses. J. Virol. 74, 7442–7450 (2000).
Rosenberg, E.S. et al. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science 278, 1447–1450 (1997).
McKay, P.F. et al. Global dysfunction of CD4 T-lymphocyte cytokine expression in simian-human immunodeficiency virus/SIV-infected monkeys is prevented by vaccination. J. Virol. 77, 4695–4702 (2003).
Reitz, M.S. Jr., Wilson, C., Naugle, C., Gallo, R.C. & Robert-Guroff, M. Generation of a neutralization-resistant variant of HIV-1 is due to selection for a point mutation in the envelope gene. Cell 54, 57–63 (1988).
Albert, J. et al. Rapid development of isolate-specific neutralizing antibodies after primary HIV-1 infection and consequent emergence of virus variants which resist neutralization by autologous sera. AIDS 4, 107–112 (1990).
Allen, T.M. et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407, 386–90 (2000).
Phillips, R.E. et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354, 453–459 (1991).
Goulder, P.J. et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3, 212–217 (1997).
Goulder, P.J. et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412, 334–338 (2001).
Barouch, D.H. et al. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415, 335–339 (2002).
Brander, C. et al. Lack of strong immune selection pressure by the immunodominant, HLA-A*0201-restricted cytotoxic T lymphocyte response in chronic human immunodeficiency virus-1 infection. J. Clin. Invest. 101, 2559–2566 (1998).
Moore, C.B. et al. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296, 1439–1443 (2002).
Yusim, K. et al. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. J. Virol. 76, 8757–8768 (2002).
Trachtenberg, E. Advantage of rare HLA supertype in HIV disease progression. Nat. Med. 9, 930–937 (2003).
Dalod, M. et al. Weak anti-HIV CD8+ T-cell effector activity in HIV primary infection. J. Clin. Invest. 104, 1431–1439 (1999).
Goulder, P.J. et al. Substantial differences in specificity of HIV-specific cytotoxic T cells in acute and chronic HIV infection. J. Exp. Med. 193, 181–194 (2001).
Meyerhans, A. et al. In vivo persistence of a HIV-1-encoded HLA-B27-restricted cytotoxic T lymphocyte epitope despite specific in vitro reactivity. Eur. J. Immunol. 21, 2637–2640 (1991).
Hay, C.M. et al. Lack of viral escape and defective in vivo activation of human immunodeficiency virus type 1-specific cytotoxic T lymphocytes in rapidly progressive infection. J. Virol. 73, 5509–5519 (1999).
Lieberman, J., Shankar, P., Manjunath, N. & Andersson, J. Dressed to kill? A review of why antiviral CD8 T lymphocytes fail to prevent progressive immunodeficiency in HIV-1 infection. Blood 98, 1667–1677 (2001).
Douek, D.C. et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature 417, 95–98 (2002).
Malhotra, U. et al. Effect of combination antiretroviral therapy on T-cell immunity in acute human immunodeficiency virus type 1 infection. J. Infect. Dis. 181, 121–131 (2000).
Oxenius, A. et al. Early highly active antiretroviral therapy for acute HIV-1 infection preserves immune function of CD8+ and CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA 97, 3382–3387 (2000).
Collins, K.L., Chen, B.K., Kalams, S.A., Walker, B.D. & Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401 (1998).
Yang, O.O. et al. Nef-mediated resistance of human immunodeficiency virus type 1 to antiviral cytotoxic T lymphocytes. J. Virol. 76, 1626–1631 (2002).
Cohen, G.B. et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10, 661–671 (1999).
Champagne, P. et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature 410, 106–111 (2001).
Migueles, S.A. et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 3, 1061–1068 (2002).
Appay, V. et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8, 379–385 (2002).
Zhang, D. et al. Most antiviral CD8 T cells during chronic viral infection do not express high levels of perforin and are not directly cytotoxic. Blood 101, 226–235 (2003).
Kostense, S. et al. Persistent numbers of tetramer+ CD8(+) T cells, but loss of interferon-gamma+ HIV-specific T cells during progression to AIDS. Blood 99, 2505–2511 (2002).
Trimble, L.A., Shankar, P., Patterson, M., Daily, J.P. & Lieberman, J. Human immunodeficiency virus-specific circulating CD8 T lymphocytes have down-modulated CD3ζ and CD28, key signaling molecules for T-cell activation. J. Virol. 74, 7320–7330 (2000).
Autran, B. et al. Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science 277, 112–116 (1997).
Ogg, G.S. et al. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J. Virol. 73, 797–800 (1999).
Luzuriaga, K. et al. Early therapy of vertical human immunodeficiency virus type 1 (HIV-1) infection: control of viral replication and absence of persistent HIV-1-specific immune responses. J. Virol. 74, 6984–6991 (2000).
Mascola, J.R. et al. Potent and synergistic neutralization of human immunodeficiency virus (HIV) type 1 primary isolates by hyperimmune anti-HIV immunoglobulin combined with monoclonal antibodies 2F5 and 2G12. J. Virol. 71, 7198–7206 (1997).
Brodie, S.J. et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat. Med. 5, 34–41 (1999).
Koenig, S. et al. Transfer of HIV-1-specific cytotoxic T lymphocytes to an AIDS patient leads to selection for mutant HIV variants and subsequent disease progression. Nat. Med. 1, 330–336 (1995).
Kovacs, J.A. et al. Increases in CD4 T lymphocytes with intermittent courses of interleukin-2 in patients with human immunodeficiency virus infection. A preliminary study. N. Engl. J. Med. 332, 567–575 (1996).
Markowitz, M. et al. Discontinuation of antiretroviral therapy commenced early during the course of human immunodeficiency virus type 1 infection, with or without adjunctive vaccination. J. Infect. Dis. 186, 634–643 (2002).
Robbins, G. et al. Augmentation of HIV-1-specific Th cell responses in chronic HIV-1 infection by therapeutic immunization. AIDS (in the press).
Lu, W., Wu, X., Lu, Y., Guo, W. & Andrieu, J.M. Therapeutic dendritic-cell vaccine for simian AIDS. Nat. Med. 9, 27–32 (2003).
Sun, J.C. & Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300, 339–342 (2003).
Shedlock, D.J. & Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300, 337–339 (2003).
Rosenberg, E.S. et al. Immune control of HIV-1 after early treatment of acute infection. Nature 407, 523–526 (2000).
Lisziewicz, J. et al. Virus control following early treatment and discontinuation of antiretroviral therapy. N. Engl. J. Med. 340, 1683–1684 (2003).
Oxenius, A. et al. Stimulation of HIV-specific cellular immunity by structured treatment interruption fails to enhance viral control in chronic HIV infection. Proc. Natl. Acad. Sci. USA 99, 13747–13752 (2002).
Altfeld, M. et al. Expansion of pre-existing, lymph node-localized CD8+ T cells during supervised treatment interruptions in chronic HIV-1 infection. J. Clin. Invest. 109, 837–843 (2002).
Garcia, F. et al. The virological and immunological consequences of structured treatment interruptions in chronic HIV-1 infection. AIDS 15, F29–F40 (2001).
Altfeld, M. et al. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J. Exp. Med. 193, 169–180 (2001).
Altfeld, M. et al. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature 420, 434–439 (2002).
Matloubian, M., Somasundaram, T., Kolhekar, S.R., Selvakumar, R. & Ahmed, R. Genetic basis of viral persistence: single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. Journal of Experimental Medicine 172, 1043–8 (1990).
Matloubian, M., Kolhekar, S.R., Somasundaram, T. & Ahmed, R. Molecular determinants of macrophage tropism and viral persistence: importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J. Virol. 67, 7340–7349 (1993).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Letvin, N., Walker, B. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat Med 9, 861–866 (2003). https://doi.org/10.1038/nm0703-861
Issue Date:
DOI: https://doi.org/10.1038/nm0703-861
This article is cited by
-
Immune surveillance for six vaccinable pathogens using paired plasma and dried blood spots in HIV infected and uninfected children in Kinshasa
Scientific Reports (2022)
-
HIV cure strategies: which ones are appropriate for Africa?
Cellular and Molecular Life Sciences (2022)
-
HIV-1 and human genetic variation
Nature Reviews Genetics (2021)
-
Beyond the replication-competent HIV reservoir: transcription and translation-competent reservoirs
Retrovirology (2018)
-
Impact of HIV-1 infection on the feto-maternal crosstalk and consequences for pregnancy outcome and infant health
Seminars in Immunopathology (2016)
