
Abstract
Cultivation-independent surveys have shown that the desert soils of Antarctica harbour surprisingly rich microbial communities1,2,3. Given that phototroph abundance varies across these Antarctic soils2,4, an enduring question is what supports life in those communities with low photosynthetic capacity3,5. Here we provide evidence that atmospheric trace gases are the primary energy sources of two Antarctic surface soil communities. We reconstructed 23 draft genomes from metagenomic reads, including genomes from the candidate bacterial phyla WPS-2 and AD3. The dominant community members encoded and expressed high-affinity hydrogenases, carbon monoxide dehydrogenases, and a RuBisCO lineage known to support chemosynthetic carbon fixation6,7. Soil microcosms aerobically scavenged atmospheric H2 and CO at rates sufficient to sustain their theoretical maintenance energy and mediated substantial levels of chemosynthetic but not photosynthetic CO2 fixation. We propose that atmospheric H2, CO2 and CO provide dependable sources of energy and carbon to support these communities, which suggests that atmospheric energy sources can provide an alternative basis for ecosystem function to solar or geological energy sources8,9. Although more extensive sampling is required to verify whether this process is widespread in terrestrial Antarctica and other oligotrophic habitats, our results provide new understanding of the minimal nutritional requirements for life and open the possibility that atmospheric gases support life on other planets.
Similar content being viewed by others


Main
Terrestrial Antarctica is among the most extreme environments on Earth. Its inhabitants experience the cumulative stresses of freezing temperatures, limited carbon, nitrogen and water availability, strong UV radiation, and frequent freeze–thaw cycles2,10,11. Although it was once believed that these conditions restrict life, we now know that the continent hosts a surprising diversity of macrofauna and microbiota1,2,12. Surveys indicate that the phylum-level composition of microbial communities in Antarctic soils is similar to those of temperate soils3, but Antarctic communities are highly specialized at the species level and strongly structured by physicochemical factors1,3,10. In many Antarctic soils, microorganisms are thought to live in dormant states2, with metabolic energy directed towards cell maintenance rather than growth13. However, it is unclear how these communities obtain the energy and carbon needed for maintenance, given that these soils are often low in organic carbon and contain few classical primary producers2,5. Cyanobacteria and algae are known primary producers in such ecosystems2,4, but multiple sites have now been described at which phototrophs are present in low abundance or restricted to lithic niches5,11,14. These findings suggest that unidentified energy and carbon sources may also support ecosystem function. Here, we used shotgun metagenomics and biochemical measurements to clarify the basis of primary production in two such Antarctic sites.
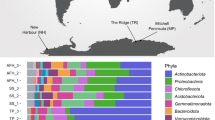
Three surface soils were initially sampled from Robinson Ridge (Wilkes Land)11 (Extended Data Fig. 1). Physicochemical analysis showed that the soils were low in total organic carbon (0.13 – 0.24%), nitrogen (0.012 – 0.023%), and moisture (4.0 – 5.6%) content (Supplementary Table 1). We used shotgun DNA sequencing to produce a 264-MB metagenome from these soils, comprising 208,233 predicted genes (Supplementary Table 2) and 451 predicted 16S/18S rRNA operational taxonomic units (OTUs) (Supplementary Table 3). The communities were dominated by Actinobacteria, Chloroflexi, Proteobacteria, Acidobacteria, and two candidate phyla, AD3 and WPS-2 (Fig. 1a). Identified phototrophs were limited to Cyanobacteria, with an average relative abundance of 0.28% (Fig. 1a). Consistent with other studies5,11,14, the low photosynthetic capacity and organic carbon content of Robinson Ridge soils contrasts with the inferred bacterial diversity, suggesting that alternative energy sources support these communities. To gain a deeper understanding of ecosystem function, we applied differential coverage binning15, which has been used to construct genomes of uncultured inhabitants of diverse environments15,16,17,18. We constructed 23 draft genomes from the Robinson Ridge metagenome, including two WPS-2 and three AD3 genomes (Fig. 1b; Supplementary Table 4). These candidate phyla affiliate with the Terrabacteria superphylum16 and are sister lineages to Chloroflexi and Armatimonadetes19 (Extended Data Fig. 2).

a, Microbial community structure of three biologically independent soil samples, as determined by mapping sequence reads onto a database of bacterial, archaeal and eukaryotic 16S/18S rRNA genes. b, Heat map showing the relative abundance of genes encoding key enzymes involved in carbon fixation and energy conservation. The relative abundance was calculated on the basis of the presence or absence of functional genes within the sequenced genomes from three biologically independent soil samples, and is presented relative to the total number of 16S rRNA gene sequences in the total microbial community.
We subsequently analysed the metabolic potential of the metagenome and derived genomes (Extended Data Fig. 3). All 23 sequenced organisms harboured terminal oxidase genes to sustain aerobic respiration, consistent with their aerated habitat, and genes for the oxidation of organic carbon compounds were also abundant (Supplementary Tables 5, 6). Genes supporting CO2 fixation through the Calvin–Benson–Bassham (CBB) cycle were unexpectedly widespread, being found in the genomes of Actinobacteria, WPS-2, and AD3 (Fig. 1b, Supplementary Table 5). Phylogenetic analysis revealed that these genomes encoded the type IE ribulose-1,5-bisphosphate carboxylase (RuBisCO) (rbcL1E)6,7 (Fig. 2b, Extended Data Fig. 4a, b); this lineage is known to support hydrogenotrophic growth of Actinobacteria7, but is absent from phototrophs. In addition, enzymes responsible for the aerobic respiration of molecular H220,21,22 and CO23,24 were encoded in multiple genomes (Fig. 1b, Supplementary Table 5). The high-affinity lineages identified—namely the structural genes encoding group 1h [NiFe]-hydrogenases (hhyL and hhyS) (Fig. 2a, Extended Data Fig. 5a) and type I [MoCu]-carbon monoxide dehydrogenases (coxL, coxS and coxM) (Extended Data Fig. 6a)—support scavenging of H2 and CO at the trace concentrations found in the atmosphere21,25. We also identified determinants of methanotrophy, ammonia oxidation, nitrogen cycling, and psychrotolerance (Supplementary Tables 5, 7–9).

Maximum-likelihood phylogenetic trees of the enzyme families responsible for atmospheric H2 respiration (a, hhyL; group 1h [NiFe] hydrogenase) and chemosynthetic CO2 fixation (b, rbcL1E; type IE RuBisCO). Protein sequences retrieved from the metagenome and reconstructed genomes were aligned against reference sequences (bootstrapped with 500 replicates).
Collectively, the metagenome data suggest that Antarctic surface soil microbial communities have the capacity to scavenge H2, CO2 and CO from the atmosphere for use as energy and carbon sources. We confirmed this theory by detecting both the expression and activity of the enzymes catalysing these processes in Robinson Ridge soils. Reverse transcription (RT)–PCR confirmed the expression of the genes encoding type IE RuBisCO (rbcL1E), high-affinity hydrogenase (hhyL), and carbon monoxide dehydrogenases (coxL) (Extended Data Fig. 7). Gas chromatography experiments showed that the soil communities aerobically oxidized atmospheric H2 and CO (threshold of 190 parts per billion by volume (p.p.b.v.) H2 and 20 p.p.b.v. CO; rate of 3.49 nmol atmospheric H2 per hour per gram dry weight and 0.42 nmol atmospheric CO per hour per gram dry weight) (Fig. 3a; Extended Data Fig. 6b; Supplementary Table 10), with H2 oxidation also measurable at −12 °C (Extended Data Fig. 5b). However, we did not observe atmospheric methane oxidation, despite recovering the genome of a putative alphaproteobacterial methanotroph. We next measured the capacity of the soils to fix carbon by tracing assimilation of 14C-labelled CO2. Although basal levels of CO2 fixation were variable (Extended Data Fig. 4c, d), the addition of H2 stimulated CO2 fixation by an average of twofold (Fig. 3b). By contrast, light stimulation had no consistent effect (Fig. 3b).

a, Gas chromatography measurements of oxidation of atmospheric H2 (mixing ratio, 0.53 parts per million by volume (p.p.m.v.)) at 10 °C. Values shown for two biologically independent soil samples (mean of technical triplicates). b, Ratios of 14C-labelled CO2 assimilation following stimulation with light and/or H2. Results shown for two biologically independent Robinson Ridge soils (technical quadruplicate) and three biologically independent Adams Flat soils (technical triplicate). Centre values show medians; boxes show upper and lower quartiles; whiskers show maximum and minimum values. Where sample size was appropriate, statistical significance between paired technical replicates was tested using a two-tailed Wilcoxon signed-rank test.
To ensure that the use of H2, CO2 and CO from the atmosphere as energy and carbon sources was not an isolated phenomenon, we sampled another Antarctic site: Adams Flat (Princess Elizabeth Land) (Extended Data Fig. 1). We collected three soils (Supplementary Table 1) with low organic carbon content and high Actinobacteria–low Cyanobacteria communities (Extended Data Fig. 8). Enzymes responsible for trace gas scavenging were expressed and active in these soils (Fig. 3a; Extended Data Figs 6b, 7; Supplementary Table 10), and H2 supplementation stimulated carbon fixation by an average of eightfold (P = 0.0039) (Fig. 3b). In further support of the theory that trace gas scavenging operates in a range of Antarctic desert sites, analysis of public metagenomes revealed that hhyL, coxL, and rbcL1E genes are relatively abundant in the McMurdo Dry Valleys region (Extended Data Fig. 9).
On the basis of these findings, we propose that the two Antarctic sites sampled harbour largely dormant microbial communities that conserve energy by oxidation of atmospheric trace gases (Fig. 4). Biochemical studies showed that soils oxidize atmospheric H2 and CO at rates that are theoretically sufficient to sustain the energy needs of their microbial communities; assuming 1.4 × 1014 cells per C-mol of biomass (the amount of biomass that contains 1 mol carbon) and a maintenance energy of 1.68 kJ per C-mol biomass per hour at 10 °C22,26, the rates of atmospheric H2 and CO oxidation observed can support 5.5 × 107 bacteria per gram of Robinson Ridge soil and 8.0 × 107 bacteria per gram of Adams Flat soil (Supplementary Table 10). Consistently, pure culture studies have shown that trace gas scavenging supports the persistence of diverse heterotrophic aerobes during organic carbon starvation20,22,25. Atmospheric H2 and CO are dependable energy sources for dormant bacteria as they are strong reductants, diffuse through cell membranes, and occur at low concentrations throughout the troposphere24,27,28.

Depicted are members of the five microbial phyla that are predicted to persist by trace gas scavenging. These phyla are variably capable of aerobic respiration of atmospheric H2 (all phyla) or CO (Actinobacteria, Dormibacteraeota), and chemosynthetic fixation of atmospheric CO2 (Actinobacteria, Dormibacteraeota, Eremiobacteraeota). It remains to be determined whether atmospheric H2 oxidation supports NAD(P)+ reduction through direct coupling or reverse electron transport, although direct coupling remains thermodynamically favourable at 10 °C (Gibbs free energy of reaction (ΔG) = −19 kJ mol−1). The community is also predicted to respire the small amount of soil organic carbon present as it becomes bioavailable.
Moreover, we propose that the primary producers in these communities are bacteria from the phyla Actinobacteria, AD3, and WPS-2 that generate biomass by consuming atmospheric H2, CO2 and CO. Of the draft genomes harbouring group 1h [NiFe]-hydrogenase genes, 55% also contained type IE RuBisCO genes (Fig. 2a, b; Supplementary Table 5), suggesting that some bacteria generate biomass through H2-driven CO2 fixation and others exclusively scavenge H2 for energy acquisition. The metagenomes suggest that minor community members may also assimilate carbon through the serine pathway (methanotrophs), the 3-hydroxypropionate pathway (ammonia oxidizers), and oxygenic photosynthesis (cyanobacteria). On the basis of the high completeness, minimal contamination, and monophyly of their genomes (Supplementary Table 4), we propose the names Candidatus Eremiobacteraeota (desert bacterial phylum) and Candidatus Dormibacteraeota (dormant bacterial phylum) for candidate phyla WPS-2 and AD3, respectively (Supplementary Information). The type genera Candidatus Eremiobacter and Candidatus Dormibacter are also proposed, based on two of the obtained genome sequences (Ga011786, Ga0137693), as per recent recommendations29.
Overall, the microbial community structure of the two Antarctic sites sampled appears to be shaped by selection for bacteria that can persist in these physically extreme, chemically deprived environments. Microbial phototrophs are in low abundance, probably restricted by climatic conditions such as low water and nutrient availability, summer radiation, and winter darkness1. Instead, most inhabitants are inferred to be dormant heterotrophic aerobic bacteria that support energy generation and, in some cases, carbon fixation by oxidizing atmospheric trace gases. In oligotrophic Antarctic soils, there is evidence that aeolian processes drive some ecosystem recruitment from less hostile microenvironments (for example, hypolithons) and niche processes subsequently select for the most functionally specialized microorganisms30; hence, the ability to scavenge trace gases is likely to be an important survival-determining trait that may influence ecosystem succession. Given the concurrent findings from Robinson Ridge, Adams Flat, and the public McMurdo Dry Valley metagenomes, trace gas scavenging may be a general mechanism in Antarctic desert soils. However, broader surveys are now needed to determine the distribution, significance, and role of this process in primary production and ecosystem succession in terrestrial Antarctica, particularly in comparison to phototrophy. Indeed, there are diverse Antarctic sites where phototrophs are dominant, including hypolithons and high-latitude or moist soils2,4. It is likely that both phototrophy and gas scavenging co-occur in many Antarctic sites, with the balance of these processes shifting depending on physicochemical factors. It will also be of interest to determine whether trace gas scavenging supports primary production in other oligotrophic ecosystems, for example the hyperarid deserts of Atacama, where Actinobacteria harbouring genes for trace gas scavenging are also dominant17. Whereas most ecosystems are driven by solar or geologically derived energy, primary production in these Antarctic desert surface soils appears to be supported by atmospheric trace gases.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Site description and sample collection
This study focused on two coastal ice-free sites in different regions of eastern Antarctica. Robinson Ridge (−66.367739, 110.585262), located 10 km south of Casey station in the Windmill Islands coast of Wilkes Land (Extended Data Fig. 1a, b), is part of a pristine polar desert (Extended Data Fig. 1c). Adams Flat (−68.5502, 78.02106) is located 242 km from Davis station in the Vestfold Hills regions of Princess Elizabeth Land (Extended Data Fig. 1d). Both sites are devoid of vascular plants, but harbour a limited diversity of macrofauna, with tardigrades and nematodes shown to be present in Robinson Ridge31. Three surface soil samples (100 g) from the top 10 cm of the soil profile were collected in December 2005 for a previous study on Robinson Ridge1 and January 2014 for Adams Flat. For both sites, samples were collected along a spatially explicit sampling design comprised of three 300-m-long transects1,11,32 separated by 2-m distances from each other. As these soils were included in the Australian Antarctic Divisions polar soil archive, samples were sieved down to 63 μm, aliquoted into 5–25-g subsamples, and stored at −80 °C until analysis.
Physicochemical analysis
Latitude, longitude, slope, aspect and elevation were recorded for each sample taken using ARC GIS software (ESRI). For all soils, detailed physical and chemical data were analysed in-house using standard procedures. Total carbon, nitrogen, and phosphorus, pH, water-holding capacity, grain size, and conductivity were recorded1. In addition, extractable ions were measured (Cl−, Br−, NO3−, NO2−, PO43−, SO42− and NH4+) and X-ray fluorescence elemental analysis was conducted (SiO2, TiO2, Al2O3, Fe2O3, MnO, MgO, CaO, Na2O, K2O, P2O5, SO3 and Cl). The soils were low in total organic carbon content (average 0.17% for Robinson Ridge, 0.09% for Adams Flat) and moisture measured as dry matter fraction (average 4.4% for Robinson Ridge, 0.42% for Adams Flat) (Supplementary Table 1).
Community DNA extraction
Total community DNA was extracted from the six soil samples for microbial community profiling and functional metagenomic analysis. In all cases, DNA was extracted from 0.25–0.3 g of each sample in technical triplicate using the FastDNA SPIN Kit for Soil (MP Biomedicals). All DNA extracts were quantified and DNA lysate quality was evaluated using automated ribosomal intergenic spacer analysis (ARISA) as described32.
Metagenome sequencing, assembly, and binning
For the three Robinson Ridge samples, DNA was extracted in triplicate and used for shotgun metagenome sequencing. Metagenome libraries were prepared using the Nextera DNA Library Preparation Kit (Illumina) and sequenced using three-fifths of an Illumina HiSeq2000 flowcell lane at the Institute for Molecular Biosciences (University of Queensland). The raw reads (2 × 100-bp reads, 14.9 Gb) were processed using Trimmomatic33 for adaptor removal and quality filtering, and BBMap to merge overlapping reads. The processed reads were combined into a large co-assembly using the de novo assembly algorithm in CLC Genomics Workbench v8 (CLC Bio) and gaps within scaffolds were closed using abyss-sealer34. Processed reads from each dataset were mapped onto the co-assembly with BamM. The scaffolds were binned on the basis of differential coverage profiles, k-mer frequencies, and GC content using GroopM35 and MetaBAT36. Population genome bins obtained with GroopM were further refined using the GroopM refine function. The genome completeness and contamination were estimated with CheckM37 by calculating the presence of lineage-specific single-copy marker genes. The two sets of population genomes from the GroopM and MetaBAT binning were compared using RefineM to identify possible duplicates. Twenty-three genomes (>50% completeness, < 10% contamination) were selected for further analysis and accounted for 56–65% of total reads obtained from the three soil samples. Of these, 11 genomes were estimated to be more than 90% complete and less than 5% contaminated (Supplementary Table 4).
Community analysis
We determined the microbial community structure of the three soil samples from Robinson Ridge and three from Adams Flat. For both sites, the bacterial and archaeal community structure was determined by 16S rRNA gene amplicon sequencing as described previously32,38. Community alpha diversity was inferred from the amplicon sequencing data by calculating observed richness, Chao1, and the Shannon index (H′) as described11. For the Robinson Ridge samples only, bacterial, archaeal, and eukaryote community structure was also determined by retrieving 16S and 18S rRNA genes from the metagenomes. Community composition profiles were generated with CommunityM by mapping reads onto a database of bacterial, archaeal and eukaryotic 16S and 18S rRNA genes (that is, the SILVA39 and GreenGenes40 databases clustered at 97% similarity). To infer the phylogeny of WPS-2 and AD3, a genome tree was generated using 38 universal conserved marker genes41 from 4,624 bacterial and archaeal genomes retrieved from the Integrated Microbial Genomes database (IMG)42 together with the recovered population genomes. A concatenated alignment of the marker genes was used to generate the genome tree with FastTree43 and the tree was visualized in iTOL44.
Functional analysis
The population genomes derived from Robinson Ridge were functionally annotated using Prokka45 and the KEGG Orthology database (Kyoto Encyclopedia of Genes and Genomes)46. Genes specifically involved in carbohydrate metabolism were identified using dbCAN, an HMM-based database for carbohydrate-active enzyme (CAZy)47 annotation. Potential secondary metabolite biosynthesis gene clusters were identified using antiSMASH48. [NiFe]-hydrogenase, [MoCu]-carbon monoxide dehydrogenase, and RuBisCO enzymes encoded within the metagenome were classified by constructing phylogenetic trees of their catalytic subunits21,49,50. The derived protein sequences encoding the catalytic subunits of these enzymes were aligned with reference sequences reported in previous studies21,49,50 using ClustalX51. Evolutionary relationships were visualized on phylogenetic trees constructed with MEGA752 using the maximum-likelihood method. Trees were bootstrapped using 500 replicates and rooted with suitable outgroup sequences. The relative abundance of hhyL, coxL, and rbcL1E in McMurdo Dry Valley samples was determined by retrieving ten public metagenomes through the Joint Genome Institute (JGI IDs 104803, 106649, 35851, 3300002548, 3300012042, 3300012045, 3300012185, 3300012188, 3300012527, and 3300012678). The genes of interest were retrieved from the downloaded metagenomes by BLAST using the hhyL, coxL, and rbcL1E gene sequences identified from the Robinson Ridge metagenome as queries. The identity of the retrieved type I coxL and type IE rbcL large subunit genes was confirmed by constructing phylogenetic trees as described above, whereas genes encoding group 1h [NiFe]-hydrogenase large subunits were identified using HydDB53. The relative abundance of the three genes was compared with those in five public forest metagenomes (JGI IDs 66726, 69782, 92543, 94443, 109646).
RT–PCR analysis
We used RT–PCR to confirm the expression of the genes of interest by the microbial communities within the six soil samples. RNA was extracted from 2 g from each of the six soil samples using the MoBio PowerSoil Total RNA Isolation kit (MO BIO). All RNA extracts were quantified using the RNA Analysis Kit (Agilent) and cDNA was synthesized using Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). RT–PCR was used to confirm the expression of the group 1h [NiFe]-hydrogenase large subunit gene hhyL (primers NiFe-244f: 5′-GGGATCTGCGGGGACAACCA-3′; NiFe-568r: 5′-TCTCCCGGGTGTAGCGGCTC-3′) and the type I [MoCu]-carbon monoxide dehydrogenase large subunit gene coxL (primers type1-1288f: 5′-TSKKYACSGGCWSSTA-3′; type1-1540r: 5′-TAYGAYWSSGGYRAYTA-3′) using previously described degenerate primers, mixtures, and thermal cycler conditions23,54. Expression of the type IE RuBisCO large subunit gene (rbcL1E) was confirmed using specifically designed primers based on the sequences obtained from Robinson Ridge (rbcL1Ef: 5′-GGACBGTSGTVTGGACSGA-3′; rbcL1Er: 5′-TTGAABCCRAAVACRTTGCC-3′). For this gene, the RT–PCR mixtures were comprised of 1× Promega reaction buffer (Promega), 0.4 mM deoxynucleotide triphosphates, 0.4 nM of each primer, 0.1 mg ml−1 bovine serum albumin, 1.25 U Go-Taq polymerase (Promega), 1 μl diluted cDNA, and nuclease-free water to a final volume of 25 μl. A standard PCR protocol was used for the type IE RuBisCO large subunit gene as follows: 95 °C for 5 min; 30 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 30 s; and a final extension of 72 °C for 5 min. Owing to the high degeneracy of coxL PCR primers, multiple bands were produced after thermal cycling. To confirm the identity of the PCR products, bands were separated by gel electrophoresis, the band of the correct size (780 bp) was excised aseptically, and its DNA was extracted using the Zymoclean Gel DNA Recovery Kit (Zymol). For hydrogenase and RuBisCO, purified PCR products were cloned and transformed using the pGEM-T Easy Vector System (Promega). The purified coxL PCR products and positive hydrogenase and RuBisCO clones were sequenced using Sanger sequencing at the Ramaciotti Center for Gene Function Analysis (Sydney). The identity of all sequenced clones was confirmed using NCBI BLAST against the nucleotide database.
Gas chromatography analysis
We used gas chromatography to determine whether the microbial communities in the collected soil samples scavenged atmospheric H2, CO, or CH4. Soil samples of 1 g were placed into sterile 114-ml serum bottles and sealed with butyl rubber stoppers. H2, CO, or CH4 gas was then added to achieve final initial headspace concentrations of ~60 p.p.m.v., ~5 p.p.m.v., or ~50 p.p.m.v., respectively. Soil incubation temperatures were monitored throughout using a 51II single input digital thermometer (Fluke). Headspaces (1 ml) were sampled in situ using a gas-tight syringe (VICI Precision Sampling). H2 and CO partial pressures were measured using a PP1 Gas Analyser (Peak Performer) equipped with a reducing compound photometer (RCP: H2, CO), flame ionizing detector (FID: CH4), Unibeads 1S 60/80 column, and Molecular Sieve 13X 60/80 column, as previously described20. Samples were calibrated against H2, CO, and CH4 standards prepared in advance. The experiments each used two biologically independent Robinson Ridge and two biologically independent Adams Flat soil samples. Gas chromatography traces were recorded of technical triplicates for all H2 measurements and technical duplicates for all CO measurements. Two negative controls, namely heat-killed soils (1 g; 121 °C, 15 p.s.i., 20 min) and serum bottles without soil, were monitored in parallel to regular sampling to ensure that the observed reduction of H2 and CO concentrations was a consequence of biological oxidation. The theoretical bacterial population sustained by trace gas scavenging (N) was calculated by the methods of Conrad26 based on the observed trace gas oxidation rate (d), the Gibbs free energy of gas oxidation (ΔG), and the theoretical maintenance energy of the population (me). ΔG was calculated to be −200.9 kJ mol−1 for the reaction H2 + 0.5 O2 → H2O and −236.2 kJ mol−1 for the reaction CO + 0.5 O2 → CO2 using the Nernst equation (10 °C, ambient gas concentrations). me was estimated to be 1.68 kJ per C-mol biomass per hour using the Tijhuis equation55.
Carbon fixation analysis
The soil samples were incubated with radiolabelled carbon dioxide (14CO2) under different conditions to determine whether their microbial communities could mediate chemosynthetic or photosynthetic CO2 fixation. Each of the soil samples (0.25 g) were added to sterile 4-ml glass vials, sealed with rubber septum lids, and stored on ice before downstream analysis. Gaseous 14CO2 (1% v/v) was generated by mixing 75 μl radiolabelled sodium bicarbonate solution (NaH14CO3, Perkin Elmer, 53.1 mCi nmol−1) with 75 μl 10% HCl solution in a sealed 4-ml glass vial and incubating for 2 h at room temperature. To each sample, 160 μl of 14CO2 (1% v/v) gas was added using a gas-tight syringe (1 ml, SGE Analytical Science), obtaining initial headspace mixing ratios of 400 p.p.m.v. 14CO2 in a headspace otherwise comprised of ambient air. In addition, 40 μl standard H2 gas (1% v/v, AirLiquide) was added to half of the sampling cohort to obtain simultaneous mixing ratios of 100 p.p.m.v. H2 and 400 p.p.m.v. 14CO2. Both groups were then incubated under either light (40 μmol photons m−2 s−1 under constant illumination) or dark conditions (sealed light-proof box) for 96 h at 10 °C. To remove any unfixed 14CO2, incubated soils were transferred to 12-ml scintillation vials, suspended in 2 ml 10% acetic acid in ethanol, and left to dry under a heat lamp at 30 °C for 12 h. Ten millilitres of scintillation cocktail (EcoLume) was added and radioisotope analysis was carried out using a liquid scintillation spectrometer (Tri-Carb 2810 TR, Perkin Elmer) operating at 95% efficiency, with background luminescence and chemiluminescence corrected through internal calibration standards. Each experiment used two Robinson Ridge soil samples in paired technical quadruplicates (performed in three separate experiments across successive weeks) and three Adams Flat samples in paired technical triplicates (performed in three separate experiments across successive weeks). Control samples were run with soils that were heat-killed for 12 h at 100 °C using a cooled vacuum drying oven (Memmert GmbH). Scintillation counts from heat-killed controls were subtracted and the amount of 14CO2 fixed per sample was calculated on the basis of the reported specific radioactivity of the original bicarbonate solution. A control experiment validated that light stimulation caused a 15-fold increase in CO2 fixation over the dark-incubated samples in a phototroph-containing soil community collected from Mitchell Peninsula in December 2005.
Data availability
The raw shotgun sequencing datasets generated during the current study have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (RR samples 1, 2 and 3 under accession numbers SRR5223441, SRR5223442 and SRR5223443, respectively). The metagenome data have been deposited into the IMG-M web portal (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) under accession number 3300009400, and the scaffolds of all 23 draft genomes have been deposited into the IMG-M web portal under accession numbers 2667527203, 2667527204 and 2698536723–2698536743).
Accession codes
References
Siciliano, S. D. et al. Soil fertility is associated with fungal and bacterial richness, whereas pH is associated with community composition in polar soil microbial communities. Soil Biol. Biochem. 78, 10–20 (2014)
Cary, S. C., McDonald, I. R., Barrett, J. E. & Cowan, D. A. On the rocks: the microbiology of Antarctic Dry Valley soils. Nat. Rev. Microbiol. 8, 129–138 (2010)
Lee, C. K., Barbier, B. A., Bottos, E. M., McDonald, I. R. & Cary, S. C. The Inter-Valley Soil Comparative Survey: the ecology of Dry Valley edaphic microbial communities. ISME J. 6, 1046–1057 (2012)
Chan, Y., Van Nostrand, J. D., Zhou, J., Pointing, S. B. & Farrell, R. L. Functional ecology of an Antarctic Dry Valley. Proc. Natl Acad. Sci. USA 110, 8990–8995 (2013)
Ji, M. et al. Microbial diversity at Mitchell Peninsula, Eastern Antarctica: a potential biodiversity “hotspot”. Polar Biol. 39, 237–249 (2015)
Tebo, B. M. et al. Microbial communities in dark oligotrophic volcanic ice cave ecosystems of Mt. Erebus, Antarctica. Front. Microbiol. 6, 179 (2015)
Grostern, A. & Alvarez-Cohen, L. RubisCO-based CO2 fixation and C1 metabolism in the actinobacterium Pseudonocardia dioxanivorans CB1190. Environ. Microbiol. 15, 3040–3053 (2013)
Kelley, D. S. et al. A serpentinite-hosted ecosystem: the Lost City hydrothermal field. Science 307, 1428–1434 (2005)
Cavicchioli, R. Microbial ecology of Antarctic aquatic systems. Nat. Rev. Microbiol. 13, 691–706 (2015)
Pointing, S. B. et al. Highly specialized microbial diversity in hyper-arid polar desert. Proc. Natl Acad. Sci. USA 106, 19964–19969 (2009)
Ferrari, B. C. et al. Geological connectivity drives microbial community structure and connectivity in polar, terrestrial ecosystems. Environ. Microbiol. 18, 1834–1849 (2016)
Chown, S. L. et al. The changing form of Antarctic biodiversity. Nature 522, 431–438 (2015)
Lennon, J. T. & Jones, S. E. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 9, 119–130 (2011)
Chanal, A. et al. The desert of Tataouine: an extreme environment that hosts a wide diversity of microorganisms and radiotolerant bacteria. Environ. Microbiol. 8, 514–525 (2006)
Albertsen, M. et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538 (2013)
Rinke, C. et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013)
Lynch, R. C., Darcy, J. L., Kane, N. C., Nemergut, D. R. & Schmidt, S. K. Metagenomic evidence for metabolism of trace atmospheric gases by high-elevation desert Actinobacteria. Front. Microbiol. 5, 698 (2014)
Howe, A. C. et al. Tackling soil diversity with the assembly of large, complex metagenomes. Proc. Natl Acad. Sci. USA 111, 4904–4909 (2014)
Lee, K. C. et al. Phylogenetic delineation of the novel phylum Armatimonadetes (former candidate division OP10) and definition of two novel candidate divisions. Appl. Environ. Microbiol. 79, 2484–2487 (2013)
Greening, C. et al. Persistence of the dominant soil phylum Acidobacteria by trace gas scavenging. Proc. Natl Acad. Sci. USA 112, 10497–10502 (2015)
Greening, C. et al. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J. 10, 761–777 (2016)
Constant, P., Chowdhury, S. P., Pratscher, J. & Conrad, R. Streptomycetes contributing to atmospheric molecular hydrogen soil uptake are widespread and encode a putative high-affinity [NiFe]-hydrogenase. Environ. Microbiol. 12, 821–829 (2010)
Quiza, L., Lalonde, I., Guertin, C. & Constant, P. Land-use influences the distribution and activity of high affinity CO-oxidizing bacteria associated to type I-coxL genotype in soil. Front. Microbiol. 5, 271 (2014)
King, G. M. & Weber, C. F. Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat. Rev. Microbiol. 5, 107–118 (2007)
Berney, M., Greening, C., Hards, K., Collins, D. & Cook, G. M. Three different [NiFe] hydrogenases confer metabolic flexibility in the obligate aerobe Mycobacterium smegmatis. Environ. Microbiol. 16, 318–330 (2014)
Conrad, R. Soil microorganisms oxidizing atmospheric trace gases (CH4, CO, H2, NO). Indian J. Microbiol. 39, 193–203 (1999)
Baani, M. & Liesack, W. Two isozymes of particulate methane monooxygenase with different methane oxidation kinetics are found in Methylocystis sp. strain SC2. Proc. Natl Acad. Sci. USA 105, 10203–10208 (2008)
Greening, C. et al. Atmospheric hydrogen scavenging: from enzymes to ecosystems. Appl. Environ. Microbiol. 81, 1190–1199 (2015)
Whitman, W. B. Modest proposals to expand the type material for naming of prokaryotes. Int. J. Syst. Evol. Microbiol. 66, 2108–2112 (2011)
Herbold, C. W., Lee, C. K., McDonald, I. R. & Cary, S. C. Evidence of global-scale aeolian dispersal and endemism in isolated geothermal microbial communities of Antarctica. Nat. Commun. 5, 3875 (2014)
Nielsen, U. N. & King, C. K. Abundance and diversity of soil invertebrates in the Windmill Islands region, East Antarctica. Polar Biol. 38, 1391–1400 (2015)
van Dorst, J. et al. Community fingerprinting in a sequencing world. FEMS Microbiol. Ecol. 89, 316–330 (2014)
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014)
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009)
Imelfort, M. et al. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ 2, e603 (2014)
Kang, D. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165 (2015)
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015)
Bissett, A. et al. Introducing BASE: the Biomes of Australian Soil Environments soil microbial diversity database. Gigascience 5, 21 (2016)
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013)
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006)
Darling, J. A., Tsai, Y. H., Blakeslee, A. M. & Roman, J. Are genes faster than crabs? Mitochondrial introgression exceeds larval dispersal during population expansion of the invasive crab Carcinus maenas. R. Soc. Open Sci. 1, 140202 (2014)
Markowitz, V. M. et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–D122 (2012)
Soo, R. M. et al. An expanded genomic representation of the phylum cyanobacteria. Genome Biol. Evol. 6, 1031–1045 (2014)
Letunic, I. & Bork, P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478 (2011)
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014)
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000)
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 (2014)
Blin, K. et al. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, W204–W212 (2013)
Park, S. W. et al. Presence of duplicate genes encoding a phylogenetically new subgroup of form I ribulose 1,5-bisphosphate carboxylase/oxygenase in Mycobacterium sp. strain JC1 DSM 3803. Res. Microbiol. 160, 159–165 (2009)
Tabita, F. R., Satagopan, S., Hanson, T. E., Kreel, N. E. & Scott, S. S. Distinct form I, II, III, and IV Rubisco proteins from the three kingdoms of life provide clues about Rubisco evolution and structure/function relationships. J. Exp. Bot. 59, 1515–1524 (2008)
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007)
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016)
Søndergaard, D., Pedersen, C. N. S. & Greening, C. HydDB: a web tool for hydrogenase classification and analysis. Sci. Rep. 6, 34212 (2016)
Constant, P., Chowdhury, S. P. & Conrad, R. Co-localization of atmospheric H2 oxidation activity and high affinity H2-oxidizing bacteria in non-axenic soil and sterile soil amended with Streptomyces sp. PCB7. Biol. Biochem. 43, 1888–1893 (2011)
Tijhuis, L., Van Loosdrecht, M. C. M. & Heijnen, J. J. A thermodynamically based correlation for maintenance Gibbs energy requirements in aerobic and anaerobic chemotrophic growth. Biotechnol. Bioeng. 42, 509–519 (1993)
Acknowledgements
We thank the Australian Antarctic Division expedition teams who obtained Antarctic soils and photos used in this study. We also thank S. Siciliano for soil sampling, D. Wilkins for the map, T. Mooney for the Adams Flat photo, M. Chuvochina and A. Oren for etymological advice, and S. Chown for valuable insights. This work was supported by Bioplatforms Australia, an Australian Antarctic Science project grant (4406; awarded to B.C.F., C.G., M.B.S. and I.S.), an ARC Future Fellowship (FT170100341; awarded to B.C.F.), an ARC DECRA Fellowship (DE170100310; awarded to C.G.), an ARC DORA and Laureate Fellowship (DP120103498 and FL150100038; awarded to P.H.), and a Marsden Grant (16-GNS-035; awarded to C.R.C. and C.G.). UNSW Sydney, the Centre for Geometric Biology (Monash University), and the Geothermal Resources of New Zealand research program (GNS Science) also provided funding to support this research.
Author information
Authors and Affiliations
Contributions
B.C.F. determined the research objective with input from all authors. I.S. provided access to soils and site information. P.H., I.V. and J.A.S. conducted metagenome sequencing and binning. M.J., B.C.F., C.G., I.V., J.v.D., J.A.S. and M.B.S. performed metagenome and genome analysis. C.G., M.J., I.V. and J.A.S. conducted phylogenetic tree inferences. M.J., J.v.D. and B.C.F. carried out RT–PCR. C.R.C., M.B.S., C.G. and K.M. carried out GC analysis. C.G., S.K.B., J.B. and T.L. conducted carbon fixation analysis. M.J., B.C.F., C.G., C.R.C., I.V., M.B.S., P.H. and S.K.B. conducted data analysis. B.C.F., C.G. and M.J. wrote the manuscript with input from all remaining authors; all authors have read and approved the manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks D. Cowan, N. Fierer, J. Schimel and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Field sites sampled in this study.
a, Map of Antarctica highlighting Casey station in the Windmill Islands and Davis station in the Vestfold Hills, eastern Antarctica. b, The location of Robinson Ridge with respect to Casey station. c, Photo of sampling site at Robinson Ridge within the Windmill Islands region. d, Photo of sampling site in Adams Flat within the Vestfold Hills region. Map courtesy of D. Wilkins; Adams Flat photo courtesy of T. Mooney.
Extended Data Figure 2 A concatenated genome tree showing phylogeny of the sequenced genomes retrieved from the Robinson Ridge metagenomes.
The phyla retrieved are coloured green. The phylogeny of the three retrieved genomes of candidate phylum Dormibacteraeota (AD3) and the two retrieved genomes of candidate phylum Eremiobacteraeota (WPS-2) is shown (coloured pink and blue, respectively). Also shown is their affiliation with the superphylum Terrabacteria (highlighted in yellow).
Extended Data Figure 3 Distribution of genes in the Robinson Ridge metagenome according to the KEGG classification system.
The most abundant metabolic categories identified were those associated with carbohydrate (18%), amino acid (17%), and energy metabolism (8%).
Extended Data Figure 4 Genetic organization, amino acid phylogeny, and inferred activity of RuBisCO enzymes in sampled soils.
a, Organization of type IE RuBisCO genes retrieved from Robinson Ridge. The genes included in Bin 12: Dormibacteraeota (AD3, top) and Bin 22: Eremiobacteraeota (WPS-2, bottom) are shown. rbcS, RuBisCO small subunit; rbcL, RuBisCO large subunit; cbbX, RuBisCO expression protein; cbbP, phosphoribulokinase; cbbR, transcriptional regulator protein; cbbT, transketolase. b, Expanded phylogenetic tree of RuBisCO large subunit genes retrieved from Robinson Ridge metagenomes. The phylogenetic tree was generated by the maximum-likelihood method and robustness was tested with 500 replicates. The majority of sequences within the metagenome and bins encode type IE RuBisCO (chemosynthesis-type), except for one of the unbinned sequences, which encodes a type IA RuBisCO (photosynthesis-type). c, Box plots showing amount of 14C-labelled CO2 assimilated by Robinson Ridge and Adams Flat soil communities following light or H2 stimulation. Results are shown for two biologically independent Robinson Ridge soils (technical quadruplicate) and three biologically independent Adams Flat soils (technical triplicate). Centre values show medians. Maxima, upper and lower quartile, and minima values are also shown. Where sample size is appropriate, statistical significance between paired technical replicates was tested using a two-tailed Wilcoxon signed-rank test. d, Amount of 14C-labelled CO2 assimilated for each individual Robinson Ridge and Adams Flat soil tested. Soils are named as in Supplementary Table 1 and results are shown for technical quadruplicates (Robinson Ridge) and technical triplicates (Adams Flat). Centre values show means, upper values show maxima, and lower values show minima. Although a variable level of basal CO2 fixation was observed, these results show that H2 addition consistently stimulated CO2 fixation.
Extended Data Figure 5 Amino acid phylogeny and inferred activity of hydrogenase enzymes in sampled soils.
a, Expanded phylogenetic tree of [NiFe]-hydrogenase large subunit genes retrieved from the Robinson Ridge metagenome data. The phylogenetic tree was generated by the maximum-likelihood method and robustness was tested with 500 replicates. Hydrogenase genes were common within the metagenome and bins. They clustered exclusively with the group 1h [NiFe]-hydrogenases, suggesting that they mediate atmospheric H2 scavenging. b, H2 oxidation activities of Robinson Ridge soils incubated at 10 °C, 4 °C, and −12 °C. Gas chromatography traces show H2 oxidation of soils in modified ambient air headspaces. Results are shown from one soil sample (Soil 1) following averaging of results from technical triplicates.
Extended Data Figure 6 Amino acid phylogeny and inferred activity of carbon monoxide dehydrogenase enzymes in sampled soils.
a, Phylogenetic tree of type I [MoCu]-hydrogenase large subunit genes retrieved from the Robinson Ridge metagenome data. The phylogenetic tree was generated by the maximum-likelihood method and robustness was tested with 500 replicates. Multiple Actinobacteria and AD3 genomes encoded this subunit, suggesting that they may mediate atmospheric CO scavenging. b, Gas chromatography results showing oxidation of atmospheric CO (mixing ratio 0.10 p.p.m.v.) mediated by the soil communities at 10 °C in modified ambient air headspaces. For both the Robinson Ridge and Adams Flat datasets, values are shown for two biologically independent soils following averaging of technical duplicates.
Extended Data Figure 7 Presence and expression of trace gas scavenging genes in Robinson Ridge and Adams Flat soil samples.
The presence and expression of the genes encoding the group 1h [NiFe]-hydrogenase large subunit (hhyL) and type IE RuBisCO large subunit (rbcL1E) was confirmed by agarose gel electrophoresis. Lane M shows the DNA ladder (Bioline 100-bp Molecular Weight Marker). Lanes 1 and 5 show amplifications from community DNA extracted from the soil samples. Lanes 2 and 6 show amplifications of cDNA derived from reverse transcription of community RNA extracted from the soil samples. No template negative controls for both the PCR (lanes 3, 7) and RT–PCR (lanes 4, 8) are also shown. Independent PCR and RT–PCR reactions were conducted twice for both Robinson Ridge and Adams Flat soil samples, with similar results. For gel source data, see Supplementary Fig. 1. For carbon monoxide dehydrogenase, the bands obtained from agarose gel electrophoresis were blurred owing to the degeneracy of the primer set. Sequencing of the bands confirmed the expression of carbon monoxide dehydrogenase genes homologous to GenBank accession numbers EU888210 (79%), EU888266 (79%), and KJ468250 (88%).
Extended Data Figure 8 Microbial community structure of Adams Flat soil samples.
Community structure was determined by targeted amplicon sequencing of bacterial 16S rRNA genes from three biologically independent soil samples. All samples show a high abundance of potential trace gas scavengers from the phylum Actinobacteria and a low abundance of phototrophs from the phylum Cyanobacteria.
Extended Data Figure 9 Comparison of the relative abundance of genes associated with trace gas scavenging in public metagenomes.
The relative abundance of the genes hhyL, coxL, and rbcL1E was compared between McMurdo Dry Valley and forest metagenomes publicly available through JGI. Results are shown for ten biologically independent McMurdo Dry Valley metagenomes and five biologically independent forest metagenomes. Centre values show medians; boxes show upper and lower quartiles; whiskers show maxima and minima. Statistical significance was tested using a one-tailed Student’s t-test.
Supplementary information
Supplementary Information
This file contains a detailed etymology of the phyla, Supplementary Tables 1-10 and Supplementary Figure 1, the uncropped scans with size marker indications. (PDF 316 kb)
Rights and permissions
The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ji, M., Greening, C., Vanwonterghem, I. et al. Atmospheric trace gases support primary production in Antarctic desert surface soil. Nature 552, 400–403 (2017). https://doi.org/10.1038/nature25014
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25014
This article is cited by
-
Biogeographic survey of soil bacterial communities across Antarctica
Microbiome (2024)
-
Trace gas oxidation sustains energy needs of a thermophilic archaeon at suboptimal temperatures
Nature Communications (2024)
-
All-inclusive nitrifiers in Antarctic soils
Nature Communications (2024)
-
Unveiling unique microbial nitrogen cycling and nitrification driver in coastal Antarctica
Nature Communications (2024)
-
Autochthonous psychrophilic hydrocarbonoclastic bacteria and its ecological function in contaminated cold environments
Biodegradation (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
