
Abstract
Over 20% of Earth’s terrestrial surface is underlain by permafrost with vast stores of carbon that, once thawed, may represent the largest future transfer of carbon from the biosphere to the atmosphere1. This process is largely dependent on microbial responses, but we know little about microbial activity in intact, let alone in thawing, permafrost. Molecular approaches have recently revealed the identities and functional gene composition of microorganisms in some permafrost soils2,3,4 and a rapid shift in functional gene composition during short-term thaw experiments3. However, the fate of permafrost carbon depends on climatic, hydrological and microbial responses to thaw at decadal scales5,6. Here we use the combination of several molecular ‘omics’ approaches to determine the phylogenetic composition of the microbial communities, including several draft genomes of novel species, their functional potential and activity in soils representing different states of thaw: intact permafrost, seasonally thawed active layer and thermokarst bog. The multi-omics strategy reveals a good correlation of process rates to omics data for dominant processes, such as methanogenesis in the bog, as well as novel survival strategies for potentially active microbes in permafrost.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
Accession codes
References
Koven, C. D. et al. Permafrost carbon-climate feedbacks accelerate global warming. Proc. Natl Acad. Sci. USA 108, 14769–14774 (2011)
Yergeau, E., Hogues, H., Whyte, L. G. & Greer, C. W. The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses. ISME J. 4, 1206–1214 (2010)
Mackelprang, R. et al. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371 (2011)
Mondav, R. et al. Discovery of a novel methanogen prevalent in thawing permafrost. Nat. Commun. 5, 3212 (2014)
Schuur, E. A. & Abbott, B. Climate change: high risk of permafrost thaw. Nature 480, 32–33 (2011)
McCalley, C. K. et al. Methane dynamics regulated by microbial community response to permafrost thaw. Nature 514, 478–481 (2014)
Jorgenson, M. T., Racine, C. H., Walters, J. C. & Osterkamp, T. E. Permafrost degradation and ecological changes associated with a warming climate in central Alaska. Clim. Change 48, 551–579 (2001)
Jansson, J. K. Towards “Tera-Terra”: terabase sequencing of terrestrial metagenomes. Microbe 6, 309–315 (2012)
Chourey, K. et al. Direct cellular lysis/protein extraction protocol for soil metaproteomics. J. Proteome Res. 9, 6615–6622 (2010)
Nicora, C. D. et al. Amino acid treatment enhances protein recovery from sediment and soils for metaproteomic studies. Proteomics 13, 2776–2785 (2013)
Auch, A. F., von Jan, M., Klenk, H. P. & Goker, M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134 (2010)
Waldrop, M. P. et al. Molecular investigations into a globally important carbon pool: permafrost-protected carbon in Alaskan soils. Glob. Change Biol. 16, 2543–2554 (2010)
Liebner, S., Harder, J. & Wagner, D. Bacterial diversity and community structure in polygonal tundra soils from Samoylov Island, Lena Delta, Siberia. Int. Microbiol. 11, 195–202 (2008)
Lovley, D. R. & Phillips, E. J. Rapid assay for microbially reducible ferric iron in aquatic sediments. Appl. Environ. Microbiol. 53, 1536–1540 (1987)
Balderston, W. L., Sherr, B. & Payne, W. J. Blockage by acetylene of nitrous oxide reduction in Pseudomonas perfectomarinus. Appl. Environ. Microbiol. 31, 504–508 (1976)
Balch, W. E., Fox, G. E., Magrum, L. J., Woese, C. R. & Wolfe, R. S. Methanogens: reevaluation of a unique biological group. Microbiol. Rev. 43, 260–296 (1979)
Zehnder, A. J. & Brock, T. D. Anaerobic methane oxidation: occurrence and ecology. Appl. Environ. Microbiol. 39, 194–204 (1980)
Macy, J. M., Snellen, J. E. & Hungate, R. E. Use of syringe methods for anaerobiosis. Am. J. Clin. Nutr. 25, 1318–1323 (1972)
Kunin, V. & Hugenholtz, P. PyroTagger: a fast, accurate pipelinde for analysis for analysis of rRNA amplicon pyrosequencing data. Open J. 1, 1–8 (2010)
Oksanen, J., Kindt, R., Legendre, P. & O’Hara, R. B. Vegan: Community Ecology Package v.1.8-2. (2006)
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336 (2010)
Markowitz, V. M. et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–D122 (2012)
Meyer, F. et al. The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386 (2008)
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson G. W CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. PeerJ PrePrints 2, e554v1 (2014)
Wu, M. & Scott, A. J. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28, 1033–1034 (2012)
Leininger, S. et al. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442, 806–809 (2006)
Giannone, R. J. et al. Proteomic characterization of cellular and molecular processes that enable the Nanoarchaeum equitans–Ignicoccus hospitalis relationship. PLoS ONE 6, e22942 (2011)
Florens, L. & Washburn, M. P. Proteomic analysis by multidimensional protein identification technology. Methods Mol. Biol. 328, 159–175 (2006)
Parks, D. H. & Beiko, R. G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721 (2010)
Meyer, D., Zeileis, A. & Hornik, K. vcd: Visualizing Categorical Data v.1.2-13. (2012)
Acknowledgements
We thank R. Hettich and the Organic and Biological Mass Spectrometry group at Oak Ridge National Laboratory for access to mass spectrometry instrumentation. M. Haw, K. Li, K. Chavarria and R. Lamendella are acknowledged for help with pre-processing frozen samples. We thank K. Billis for help with RNA sequence preprocessing. This work was partly supported by the Director, Office of Science, Office of Biological and Environmental Research, Climate and Environmental Science Division, of the US Department of Energy, Terrestrial Ecosystem Science-Scientific Focus Area (TES-SFA), through a Community Sequencing Project at the DOE Joint Genome Institute (JGI CSP - 152) and by a Lawrence Berkeley National Laboratory Laboratory Directed Research & Development (LDRD) grant, all under contract number DE-AC02-05CH11231; by the Pacific Northwest National Laboratory under contract number DE-AC05-76RL01830; and by the Danish National Research Foundation (CENPERM DNRF100). The work conducted by the US Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the US Department of Energy under contract number DE-AC02-05CH11231. Additional funding and considerable logistic support were provided by the Bonanza Creek Long-Term Ecological Research Program, which is jointly funded by National Science Foundation (DEB 1026415) and the US Department of Agriculture Forest Service, Pacific Northwest Research Station (PNW01-JV112619320-16). Support was also received from the US Geological Survey Climate R&D Program and Alaska Climate Science Center. J.Hu. was supported by Academy of Finland grant number 135669.
Author information
Authors and Affiliations
Contributions
J.K.J., M.P.W. and J.Hu. planned the study. M.P.W. and J.M. collected the samples and J.M. performed the chemical analyses. J.Ha., M.R.T. and A.D.M. performed the site characterization. M.B.S., N.C.V., M.M.D. and L.H.L. performed the proteomics. J.Hu., R.M., S.J.B. and K.M. analysed the sequence data. J.Hu., M.P.W. and J.K.J. wrote the paper with contributions from all authors. J.Hu., J.K.J., M.P.W. and R.M. made the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
The 454 nucleotide sequences have been deposited in Sequence Read Archive under BioProject number PRJNA222786, metagenomes and metatranscriptomes in IMG/M under Study ID Gs0063124 MG-RAST under project number 11953 and proteomics data in the PRIDE partner repository under dataset identifier PXD001131.
Extended data figures and tables
Extended Data Figure 1 Comparison of multi-omics data between the three zones.
The triplots visually present selected functions in (a) metagenomes, (b) metatranscriptomes and (c) metaproteomes in the three studied zones (permafrost, active layer and thermokarst bog). The colours correspond to SEED subsystems categories in MG-RAST; in addition, the closer the symbol is to the node of the triplot, the more abundant the gene, transcript or protein is in that particular soil type. Functions in the centre are shared among all three sites, those on edges are shared between two zones and those on the nodes are unique to that site. Owing to the large number of shared functions between bog and active layer in the metatranscriptomes, these categories are specifically compared in d.
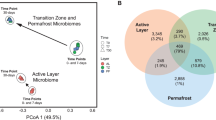
Extended Data Figure 2 Shared and unique genes, transcripts and proteins in the three zones.
The Venn diagrams for (a) metagenome, (b) metatranscriptome and (c) metaproteome data illustrate the shared and unique genes, transcripts and proteins in the three zones.
Extended Data Figure 4 Abundance of genes involved in sulphate reduction, nitrogen cycle, methanogenesis and methane oxidation.
The relative abundance of each gene is shown; error bars, s.d. The genes with protein matches in the metaproteome data are marked with a triangle.
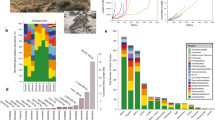
Extended Data Figure 5 The 20 most abundant OTUs found in permafrost, active layer and thermokarst bog.
a, Bacterial OTUs; b, archaeal OTUs. A single Chloroflexi OTU constituted up to 15% of the OTUs in permafrost and a draft genome bin was obtained that corresponded to this OTU (Extended Data Table 1). The abundant archaeal OTUs were methanogens.
Extended Data Figure 6 Comparison of abundant KEGG orthologous groups found in all three omics approaches (MG, MT and MP) for each site.
Permafrost: restriction modification systems, PAS sensors, ABC transporters for oligopeptides and nucleic-acid synthesis. Active layer: chaperonins, dehydrogenases and transporters for branched-chain amino acids and sugars. Thermokarst bog: methyl coenzyme M reductase, glycerol kinase, nucleic-acid synthesis and ATPases.
Supplementary information
Supplementary Information
This file contains Supplementary Text and references. (PDF 184 kb)
Supplementary Data
This file contains Supplementary Table 1. (XLSX 190 kb)
Supplementary Data
This file contains Supplementary Table 2. (XLSX 13 kb)
Supplementary Data
This file contains Supplementary Table 3. (XLSX 200 kb)
Rights and permissions
About this article

Cite this article
Hultman, J., Waldrop, M., Mackelprang, R. et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 521, 208–212 (2015). https://doi.org/10.1038/nature14238
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14238
This article is cited by
-
Weathered granites and soils harbour microbes with lanthanide-dependent methylotrophic enzymes
BMC Biology (2024)
-
Structure and ecological function of the soil microbiome associated with ‘Sanghuang’ mushrooms suffering from fungal diseases
BMC Microbiology (2023)
-
Microbial life in 25-m-deep boreholes in ancient permafrost illuminated by metagenomics
Environmental Microbiome (2023)
-
Soil viral diversity, ecology and climate change
Nature Reviews Microbiology (2023)
-
Genomic evidence that microbial carbon degradation is dominated by iron redox metabolism in thawing permafrost
ISME Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
