
Abstract
Malignant rhabdoid tumor has traditionally been defined by its histologic phenotype. However, genetic investigations of malignant rhabdoid tumor have revealed a characteristic loss of or mutation in the INI1 gene on chromosome 22q. The occurrence and significance of soft tissue tumors meeting genetic criteria for malignant rhabdoid tumor but with an undifferentiated non-rhabdoid histology is poorly characterized. Seventeen undifferentiated sarcomas, lacking rhabdoid histology were identified either through the surgical pathology files of The Children's Hospital of Philadelphia (1980–2005) or in consultation. Immunohistochemistry for the INI1 protein showed a loss of nuclear expression within tumor cells in five of these cases. On histologic review, these five tumors had a featureless sheet-like architecture; four were small round blue cell tumors, and one showed focal spindling. Although they had variably prominent nucleoli, classic rhabdoid morphologic features were not identified in any of these cases at primary presentation. Additional immunohistochemistry showed a polyphenotypic profile. Four of the five tumors showed genetic abnormalities involving the INI1 gene by a combination of fluorescent in situ hybridization, reverse transcription-polymerase chain reaction, and/or mutational analysis. Patient ages ranged from 1 week to 5 years. Four patients were male, and one was female. Sites included two neck tumors, two extremity tumors, and one paraspinal tumor. Two patients are alive and well over 15 years from the time of diagnosis; the remaining four are alive and well but with less than 2 years follow-up. Thus, alterations of the INI1 gene with consequent loss of expression identified a population of undifferentiated sarcomas lacking classic rhabdoid morphology in young patients, with evidence of favorable survival. Whether these undifferentiated sarcomas represent a clinicopathologic entity distinct from classic malignant rhabdoid tumor requires further investigation.
Similar content being viewed by others


Main
Malignant rhabdoid tumor is a rare neoplasm originally described in the kidney,1 but subsequently identified in a large number of extrarenal locations, particularly soft tissues, and the central nervous system, where it is more commonly referred to as atypical teratoid/rhabdoid tumor.2 It is a predominantly pediatric neoplasm, with most tumors occurring in infants and young children. Malignant rhabdoid tumors are aggressive neoplasms that demonstrate a poor response to treatment and are typically rapidly fatal.3
These tumors have traditionally been defined by their characteristic light microscopic appearance. Malignant rhabdoid tumors tend to grow in sheets, but may also show a trabecular, alveolar, or discohesive pattern. The classic rhabdoid tumor cell appears large with an eccentrically placed nucleus with vesicular chromatin and a prominent eosinophilic nucleolus. These cells characteristically have abundant eosinophilic cytoplasm with a globular pink cytoplasmic inclusion, which corresponds to whorled intermediate filaments as seen by electron microscopy.4 Molecular genetic investigations of malignant rhabdoid tumors have identified a characteristic loss or mutation of the INI1 (also known as SNF5, SMARCB1, BAF47) gene in chromosome band 22q11.2.5, 6 INI1 is part of the SWI/SNF chromatin remodeling complex, which acts as both a transcriptional repressor and activator and is constitutively expressed in all cells. Germline mutations or deletions of INI1 predispose patients to the development of rhabdoid tumors, and homozygous inactivation of INI1 in human tumors support its role as a tumor suppressor gene.6 Deletion and/or mutation of both copies of the INI1 gene results in loss of INI1 expression at the protein level, which can be detected using immunohistochemistry with an anti-INI1 antibody.7, 8, 9 INI1 immunohistochemistry has been demonstrated to be sensitive and specific for the diagnosis of malignant rhabdoid tumor, and INI1 protein loss appears to be quite rare in other tumors.7, 8, 9
Occasional malignant tumors encountered in soft tissue lack any identifiable clues as to histogenesis and even after extensive investigations are best classified as ‘undifferentiated.’ Whether such undifferentiated neoplasms might demonstrate loss of INI1 protein expression at the immunohistochemical level and harbor underlying INI1 gene abnormalities, despite absence of typical rhabdoid histologic features is unknown. Thus, the aim of this study was to identify and determine the significance of tumors with INI1 protein loss but without evidence of rhabdoid morphology by histologic evaluation.
Materials and methods
This study was conducted with the approval of the Institutional Review Board of The Children's Hospital of Philadelphia. The surgical and consultation pathology files of the hospital were searched for cases arising in soft tissue diagnosed as undifferentiated sarcoma between 1980 and 2005. Routine hematoxylin-eosin-stained sections from each case were reviewed by two pathologists (BRP and PAK). Cases were selected for further study if there was adequate tissue for light microscopy and immunohistochemistry, a lack of histologic differentiation, including a total absence of classic light microscopic rhabdoid features in any area of the primary tumor, and failure of immunohistochemical analysis, other than BAF47/SNF5, to establish a specific histologic diagnosis.
For the selected cases, immunohistochemistry on formalin-fixed, paraffin-embedded tissues was performed for the INI1 protein using previously published methods.8 Briefly, the BAF47/SNF5 mouse monoclonal antibody (BD Transduction Labs, San Diego, CA, USA) was utilized with a DAKO autostainer® (Dako, Carpinteria, CA, USA). Following heat-induced antigen retrieval in citrate buffer, sections were incubated with the primary antibody at 1:40 dilution for 30 min at room temperature. Detection utilized the DAKO Envision Plus HRP secondary anti-mouse antibody and DAB, and the slides were counterstained with hematoxylin. Additional immunohistochemistry was performed on the selected cases with adequate controls for the following: AE1/AE3 cytokeratins (Dako), epithelial membrane antigen (EMA; Dako), CD99 (MIC2 clone; DAKO), vimentin (Dako), smooth muscle actin (SMA; Dako), muscle specific actin (MSA; Enzo, New York, NY, USA), S100 protein (Dako), synaptophysin (Boehringer Mannheim, Basel, Switzerland), neuron specific enolase (NSE; Dako), CD34 (Biogenex, San Ramon, CA, USA), and desmin (Dako). For CD99, only strong linear perimembranous staining was interpreted as positive. For smooth muscle actin, staining was only considered positive if the intensity was comparable to the internal blood vessel controls.
Tumors which did not express INI1 by immunohistochemistry were also evaluated by fluorescence in situ hybridization, reverse transcription-polymerase chain reaction, and/or direct sequencing for deletion or mutation of the INI1 gene using previously published methods.10 Loss of expression by reverse transcription-polymerase chain reaction in the absence of a coding sequence mutation was considered a positive result for gene inactivation.
Electron microscopy was performed in three of the selected cases. Molecular analysis using reverse transcription-polymerase chain reaction for known sarcoma-associated fusion products had been performed at the time of original diagnosis in two cases. Clinical information was obtained by chart review.
Results
A total of 18 cases originally diagnosed as undifferentiated sarcomas were identified through review of departmental records. Histologic review for this study demonstrated focal rhabdoid features in one of these cases, which was excluded from further study, leaving 17 cases. Of these, 13 had adequate material for immunohistochemistry. INI1 was not retained in five cases; the clinical features of these cases are detailed in Table 1. There were two infants and three young children, ranging in age from 1 week to 5 years, 1 month. Four patients were male, and one was female. Primary tumor sites included: neck (two cases), extremities (two cases), and spinal epidural space (one case). Three patients had metastatic disease at diagnosis: one each in marrow, pelvis, and lung. Despite extensive pathologic work up including immunohistochemistry and in three cases electron microscopy, a specific pathologic diagnosis at time of presentation beyond that of undifferentiated sarcoma could not be established. In one case (case 3), the original diagnosis of undifferentiated sarcoma had been revised to embryonal rhabdomyosarcoma based on central review; however, repeated immunohistochemistry failed to reveal convincing positivity for either desmin or muscle specific actin. With the exception of case 1, where much of the treatment regimen is unknown, all patients were treated by surgical excision followed by chemotherapy. Patients 3 and 5 also received radiation therapy. Only one of the five patients is known to have had a recurrence, which was local, and all of the patients are alive and free of disease with variable lengths of follow-up ranging up to 21 years.
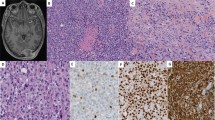
The histologic features of the tumors are outlined in Table 2 and illustrated in Figures 1 and 2. Although subtle differences were appreciated, all had a predominantly solid growth pattern with the tumor cells arrayed in patternless sheets, and with variable cellular discohesion. In two cases there was a perivascular growth pattern imparted by extensive tumoral necrosis. A nested to trabecular pattern was apparent in regions of case 5, and extensive necrosis was present in case 2. Four of the tumors (cases 2–5) showed a small round blue cell tumor morphology with the presence of minimal to moderate cytoplasm; one case (case 1) had slight focal spindling of tumor cells. Two cases (cases 2 and 3) had unusually prominent gaping blood vessels. None of the cases at diagnosis had rhabdoid-type glassy cytoplasmic inclusions, and none had associated nuclear indentation, although it is interesting to note that at definitive surgery following chemotherapy, one case (case 4) displayed classic rhabdoid morphologic features. All cases had numerous mitoses and numerous apoptotic cells. Nuclear features were varied; three cases had vesicular nuclei, and all had variable numbers of moderately prominent nucleoli. In fact, in case 1, the presence of vesicular nuclei and moderately prominent nucleoli was sufficient to raise the possibility of malignant rhabdoid tumor in the differential diagnosis, however classic rhabdoid pink globular cytoplasmic inclusions with associated nuclear indentation were not apparent in any microscopic fields.

Low power photomicrographs demonstrating two patterns of growth seen in undifferentiated sarcomas lacking INI1 expression. (a) Diffuse growth pattern with tumor cells arranged around gaping blood vessel, associated with peripheral necrosis. (b) A nested pattern composed of trabeculae of small round tumor cells (H&E, original magnification × 100).

High power photomicrographs of undifferentiated sarcomas lacking INI1 expression, showing round to slightly spindled tumor cells with ill-defined cytoplasm and variably prominent nucleoli, and representative INI1 immunohistochemical stain showing loss of expression in tumor cells with retention of nuclear staining in endothelial cells and inflammatory cells. (Original magnification × 400.)
Consistent with the inclusion criteria for this study, all tumors showed complete loss of nuclear INI1 expression by immunohistochemistry (see Figure 2), with retained expression within vascular endothelia and infiltrating inflammatory cells serving as internal positive controls. Additional immunohistochemistry (Table 3) showed a polyphenotypic profile as follows: vimentin+[5/5], cytokeratin+[5/5], EMA+[4/5], perimembranous CD99+[2/5], SMA+[3/5], MSA+[3/4], S100+[2/5], NSE+[2/4], CD34+[1/4], synaptophysin+[0/4], desmin+[0/4]. Globular, ‘rhabdoid’ type inclusions were not identified with either cytokeratin or vimentin stains in any of the cases.
Electron microscopy was performed at the time of original diagnosis in three of six cases (cases 2, 4, and 5), and in all three cases ultrastructural examination (Figure 3) demonstrated undifferentiated tumor cells, with many of the cells having prominent nucleoli, but with no evidence of intracytoplasmic inclusions composed of whorled intermediate filaments. Most cells had relatively little cytoplasm. Neuritic processes and dense core granules were not seen, and basal lamina and intercellular junctions were not identified.

Representative electron photomicrograph of Case 4, biopsy pretreatment: Tumor cells with large nuclei, prominent nucleoli, and relatively little cytoplasm, with few organelles. Aggregates of cytoplasmic filaments are not seen within the cytoplasm, nor are there any distinguishing features to subclassify this tumor ultrastructurally beyond an undifferentiated sarcoma.
Genetic studies (Table 4) revealed abnormalities of the INI1 gene in four cases; one of the cases could not be evaluated due to insufficient material (case 2). Specifically, two cases demonstrated homozygous deletion of the INI1 gene by fluorescent in situ hybridization (cases 3 and 4). Case 1 demonstrated a classic mutation in exon 5 of the INI1 gene, whereas reverse transcription-polymerase chain reaction analysis of case 5 showed low expression of the INI1 gene indicative of an INI1 gene abnormality. Fusion analyses by reverse transcription-polymerase chain reaction, performed in cases 4 and 5 were negative for the fusion products of alveolar rhabdomyosarcoma, Ewing's sarcoma, and synovial sarcoma; case 5 was additionally negative for the fusion product of desmoplastic small round cell tumor.
Discussion
Soft tissue sarcomas constitute 7% of malignancies in children 15 years and younger, with approximately 600 new cases diagnosed in the United States every year.11 The great majority of these can be placed into well classified diagnostic groups, and the recent advent of readily available molecular testing as an adjunct to histologic analysis, immunohistochemistry, and electron microscopy has contributed to accurate and confident diagnosis in an increasing percentage of cases. More than half of pediatric soft tissue sarcomas are rhabdomyosarcomas or Ewing's sarcoma/primitive neuroectodermal tumor, and most other frankly malignant soft tissue sarcomas fall into the categories of malignant peripheral nerve sheath tumors, epithelioid sarcomas, fibrosarcomas, synovial sarcomas, and desmoplastic small round cell tumors. However, there remain a small group of pediatric sarcomas, which are unclassifiable and are referred to as undifferentiated soft tissue sarcomas. As there is no well established histologic or immunohistochemical profile for undifferentiated soft tissue sarcomas, and the diagnosis also rests upon the absence of any reproducible molecular genetic aberration, the diagnosis of undifferentiated soft tissue sarcoma is never one of certainty, and is considered generally to be a diagnosis of exclusion. These are a rare group of tumors, as attested by the fact that only 8% of pediatric sarcomas in the IRS-III and IRS IV pilot series were classified as undifferentiated soft tissue sarcomas.12 Given continuing advances in immunohistochemistry and molecular testing, it is very likely that the true incidence of these tumors is actually lower.
Histologically, most cases of undifferentiated soft tissue sarcoma are characterized by formless sheets of packed round to vaguely spindled cells, similar to the cases in this series. Apart from near universal vimentin expression, pediatric undifferentiated soft tissue sarcomas have not shown a consistent immunohistochemical profile. Somers et al13 noted that 4–15% of undifferentiated soft tissue sarcomas display polyphenotypic immunohistochemical findings, and in our series, the presence of a polyphenotypic immunostaining profile marked these cases as worthy of further investigation. However, these studies did not support a more definitive diagnosis in any case beyond that of undifferentiated soft tissue sarcoma. Because of the relatively young age of the patients in this series, as well as the reported occurrence of loss of INI1 expression in some epithelioid sarcomas and weak or focal expression in some synovial sarcomas, INI1 expression was evaluated in these cases.7 The observation of loss of INI1 expression was further confirmed in 4/5 cases by identifying alterations of the INI1 gene through a combination of techniques including sequence analysis, reverse transcription-polymerase chain reaction, and fluorescent in-situ hybridization.
The differential diagnosis for our cases includes classic epithelioid sarcoma, proximal-type epithelioid sarcoma, synovial sarcoma, desmoplastic small round cell tumor, Ewing's sarcoma and malignant rhabdoid tumor. Classic epithelioid sarcoma, a tumor that expresses vimentin, cytokeratin, and epithelial membrane antigen would be unlikely, given the young age of our patients; moreover, only one of these tumors arose in the distal extremity, and none demonstrated epithelioid cytology or necrobiotic nodules. Likewise, monophasic synovial sarcoma typically displays a uniform spindle cell morphology, which was not apparent in any of these tumors. Although synovial sarcoma would display vimentin and epithelial markers, the presence of muscle markers in several of our cases would also be inconsistent with synovial sarcoma. Finally, presence of the SSX–SYT fusion protein was specifically excluded in cases 4 and 5. Desmoplastic small round cell tumor, although polyphenotypic, generally expresses desmin, and none of our cases were desmin positive. None of our cases displayed significant desmoplasia, and as in the case of epithelioid sarcoma, the young age of our patients would be unusual for desmoplastic small round cell tumor. Ewing's sarcoma could be confidently ruled out in four of these cases on molecular genetic and immunohistochemical grounds, as these cases either failed to stain for CD99 or lacked an EWS related fusion. Ewing's sarcoma could not be absolutely excluded in case 3, as this tumor expressed CD99 in a membranous fashion focally, and molecular results were not available. However, the histologic findings of vesicular nuclei, moderately prominent nucleoli, and the expression of muscle antigens would be unlikely for Ewing's sarcoma.
The most problematic tumors in the differential diagnosis of these cases are the proximal-type epithelioid sarcomas and malignant rhabdoid tumor. The proximal variant of epithelioid sarcoma is a recently described, deep-seated, and aggressive malignancy of soft tissue, with a predilection for involvement of the pelvic and genital tract regions.14 There is extensive histologic and immunophenotypic overlap with malignant rhabdoid tumor, and recent molecular studies have shown that inactivation of the INI1 gene is frequently present in these tumors,15 suggesting a very close relationship to malignant rhabdoid tumor in at least some cases. Although morphologically similar to the tumors described as proximal variants of epithelioid sarcoma, our cases occurred in a younger age group, and none in the pelvic/genitourinary region. Also, reactivity for CD34, which is positive in approximately 50% of epithelioid sarcomas, was only seen in one of these tumors (case 4).
It is well recognized that both central nervous system atypical teratoid/rhaboid tumors and malignant rhabdoid tumors of soft tissues may have focal or extensive areas lacking rhabdoid features. However, under current histologic criteria, a diagnosis of malignant rhabdoid tumor cannot be made without at least focal evidence of rhabdoid cells, which contain prominent globoid, hyaline eosinophilic cytoplasmic inclusions. At diagnosis, all of our cases were totally lacking such cells, despite the availability of abundant lesional tissue for examination. It can be argued that some of these cases could be rhabdoid tumors in which the diagnostic morphologic features were either very focal or absent. Indeed, in case 4, following chemotherapy, the definitive surgical specimen did show classic morphology of malignant rhabdoid tumor, lending plausibility to such an interpretation. What is unusual is that none of the tumors in this series has demonstrated the poor clinical outcome typical of malignant rhabdoid tumor; rather, to date all patients are alive and well. Short follow-up times in several cases suggest caution in this conclusion, but generally the outcomes for these patients were more favorable than other cases of pediatric undifferentiated sarcoma that have been reported, and certainly more favorable than for malignant rhabdoid tumor.
With the exception of case 4, in none of these cases would a diagnosis of malignant rhabdoid tumor have been considered in the absence of loss of nuclear expression of INI1. Loss of INI1 expression, as determined by immunohistochemistry is relatively specific for renal and extrarenal malignant rhabdoid tumor.7, 8, 9 Hoot et al and Sigauke et al found a loss of nuclear INI1 expression in a combined total of 10 extrarenal malignant rhabdoid tumors and 22 renal malignant rhabdoid tumors. Conversely, retained expression was seen in Ewing's sarcoma, alveolar and embryonal rhabdomyosarcoma, desmoplastic small round cell tumor, clear cell sarcoma (both renal and extrarenal), Wilms' tumor, X-linked renal cell carcinoma, and cellular congenital mesoblastic nephroma.7, 9 Variable expression was reported by Hoot et al for synovial and epithelioid sarcomas; Sigauke et al found loss of expression in epithelioid sarcomas and renal medullary carcinoma. Cheng et al16 found loss of INI1 expression in all five renal medullary carcinomas they examined, and correlated this with aggressive behavior. The two undifferentiated sarcomas in the study by Hoot et al7 showed retained INI1 expression, and moreover, none of the 13 cases of undifferentiated soft tissue sarcomas studied by Somers et al13 demonstrated loss of INI1 expression. However, the abnormal molecular genetic results in four of the five tumors in our series lend additional support to the role of loss of INI1 expression in some of these tumors.
Most tumors with loss of INI1 expression are associated with aggressive biologic behavior and poor clinical outcomes. The opportunity to query INI1 status in a wide range of central nervous system, renal and soft tissue tumors has been facilitated by the recent development of reliable immunohistochemical staining techniques for this protein.8, 17 Although functioning as a tumor suppressor gene, the exact role of INI1 in tumorigenesis is not well understood. However it is possible that the loss of INI1 expression may be associated with a range of biologic behaviors in tumors. This possibility is further strengthened by the rare occurrence of low grade or benign central nervous system tumors which undergo delayed malignant progression and subsequent loss of INI1 expression.18, 19 In conjunction with the reports of loss of INI1 expression in some epithelioid sarcomas as well as renal medullary carcinomas, these results suggest that loss of INI1 expression may be a biologic event of uncertain prognostic significance both in tumors which have rhabdoid histologic features, and in tumors which lack a well-defined rhabdoid phenotype. In the cases reported here, it appears that INI1 expression may be a defining feature of these tumors whereas not necessarily conferring the poor prognosis typical of malignant rhabdoid tumors. A further complication lies in the fact that the exact nature of the relationship between some of the tumors in which loss of INI1 expression has been identified (including epithelioid sarcomas and soft tissue malignant rhabdoid tumors) has yet to definitively established, making it difficult to draw broad conclusions in the absence of further larger studies.
These findings highlight that within undifferentiated sarcomas there is a subset of tumors that can be uniquely defined by the loss of INI1 protein associated with mutation, deletion, or low expression of INI1. Histologically these tumors are characterized by poorly differentiated small round to spindled cells with a polyphenotypic immunostaining profile including muscle and neural (neuron-specific enolase) expression. The clinical outcomes in these cases appear to be more favorable than those encountered in malignant rhabdoid tumor, although the numbers of cases are too small to draw a firm conclusion. Although further study of additional cases is required to determine if these represent a unique entity, the clinical outcomes in this series suggest a more favorable prognosis than other tumors that would be considered in the differential diagnosis. Thus, we recommend that INI1 immunohistochemical staining should be considered in the evaluation of undifferentiated sarcomas for both diagnostic and prognostic purposes.
References
Beckwith JB, Palmer NF . Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer 1978;41:1937–1948.
Rorke LB, Packer RJ, Biegel JA . Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 1996;85:56–65.
Oda Y, Tsuneyoshi M . Extrarenal rhabdoid tumors of soft tissue: clinicopathological and molecular genetic review and distinction from other soft-tissue sarcomas with rhabdoid features. Pathol Int 2006;56:287–295.
Schofield D . Extrarenal rhabdoid tumor. In: Fletcher CDM, Unni KK, Mertens F (eds). World Health Organization Classification of Tumours: Patholgy and Genetics of Soft tissue and Bone. IARC Press: Lyon, 2002, pp 219–222.
Biegel JA, Allen CS, Kawasaki K, et al. Narrowing the critical region for a rhabdoid tumor locus in 22q11. Genes Chromosomes Cancer 1996;16:94–105.
Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998;394:203–206.
Hoot AC, Russo P, Judkins AR, et al. Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol 2004;28:1485–1491.
Judkins AR, Mauger J, Ht A, et al. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 2004;28:644–650.
Sigauke E, Rakheja D, Maddox DL, et al. Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: an immunohistochemical study with implications for diagnosis. Mod Pathol 2006;19:717–725.
Perry A, Fuller CE, Judkins AR, et al. INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas. Mod Pathol 2005;18:951–958.
Dehner LP . Clinicopathologic aspects of soft tissue tumors. In: Coffin CM, Dehner LP, O'Shea PA (eds). Pediatric Soft Tissue Tumors: A Clincal, Pathological, and Therapeutic Approach. Williams and Wilkins: Baltimore, 1997, pp 6–7.
Pawel BR, Hamoudi AB, Asmar L, et al. Undifferentiated sarcomas of children: pathology and clinical behavior-an Intergroup Rhabdomyosarcoma study. Med Pediatr Oncol 1997;29:170–180.
Somers GR, Gupta AA, Doria AS, et al. Pediatric undifferentiated sarcoma of the soft tissues: a clinicopathologic study. Pediatr Dev Pathol 2006;9:132–142.
Guillou LKY . Epithelioid sarcoma. In: Fletcher CDM, Unni KK, Mertens F (eds). World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press: Lyon, 2002, pp 205–207.
Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 2005;65:4012–4019.
Cheng JX, Tretiakova M, Gong C, et al. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol 2008;21:647–652.
Judkins AR . Immunohistochemistry of INI1 expression: a new tool for old challenges in CNS and soft tissue pathology. Adv Anat Pathol 2007;14:335–339.
Allen JC, Judkins AR, Rosenblum MK, et al. Atypical teratoid/rhabdoid tumor evolving from an optic pathway ganglioglioma: case study. Neuro Oncol 2006;8:79–82.
Chacko G, Chacko AG, Dunham CP, et al. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neurooncol 2007;84:217–222.
Acknowledgements
We acknowledge Dr Chi K Nguyen of Pensacola Pathologists (Pensacola, FL, USA) for furnishing Case 5 in consultation. We thank Luanne Wainwright and Lu Tan for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
The authors declare that there are no conflicts of interest to disclose.
Rights and permissions
About this article
Cite this article
Kreiger, P., Judkins, A., Russo, P. et al. Loss of INI1 expression defines a unique subset of pediatric undifferentiated soft tissue sarcomas. Mod Pathol 22, 142–150 (2009). https://doi.org/10.1038/modpathol.2008.185
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2008.185
Keywords
This article is cited by
-
Proximal-Type Epithelioid Sarcoma in Skull Base: a Pathological Diagnosis Challenge with Other Intracranial Tumors
Pathology & Oncology Research (2019)
-
Marked for death: targeting epigenetic changes in cancer
Nature Reviews Drug Discovery (2017)
-
An Xp11.2 translocation renal cell carcinoma with SMARCB1 (INI1) inactivation in adult end-stage renal disease: a case report
Diagnostic Pathology (2016)
-
Reclassification of rhabdoid tumor and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression: three subtypes of rhabdoid tumor according to their histological features
Modern Pathology (2016)
-
Driver mutations of cancer epigenomes
Protein & Cell (2014)
