
Abstract
The paper presents the principles and the results of the implementation of dielectrophoresis for separation and identification of rare cells such as circulation tumor cells (CTCs) from diluted blood specimens in media and further label-free identification of the origins of separated cells using radio-frequency (RF) imaging. The separation and the identification units use same fabrication methods which enable system integration on the same platform. The designs use the advantage of higher surface volume ratio which represents the particular feature for micro- and nanotechnologies. Diluted blood in solution of sucrose–dextrose 1–10 is used for cell separation that yields more than 95.3% efficiency. For enhanced sensitivity in identification, RF imaging is performed in 3.5–1 solution of glycerol and trypsin. Resonance cavity performance method is used to determine the constant permittivity of the cell lines. The results illustrated by the signature of specific cells subjected to RF imaging suggest a reliable label-free single cell detection method for identification of the type of CTC.
Similar content being viewed by others

Introduction
Over the last few decades, medical research has gained significant evolvement that has gently led toward a paradigm shift approach in medicine: moving the observation from patient and organ toward a more in-depth observation—single cells within the organ. This advancement has been enabled to some extent by engineering sciences, among which a major momentum was driven by the microsystems technologies.
The micro- and nanotechnologies have been research subjects of the past few decades by engineers and scientists. Some of the investigations on microsystems have been directed toward the applications on medical sciences which enabled promising progress of few research topics in medicine1. Thus, at micro scale, elements of interest to medical sciences existent in the specific investigated matter could be revealed, analyzed, and the results could lead the track toward procedures that could not have been imagined two decades ago2. As a good example to the above statement, single living cell analysis has the potential to help in early detection of medical conditions involving modifications in the cell functions such as cancer genesis and progression3.
The presented research was inspired by the need to accurately count circulating tumor cells during and after the treatment of cancer patients3,4. An added benefit to the process could be offered by the capability that the counted cells could be collected for further analysis of culturing after counting and identification. Hence, the rationale of the research resides in achieving label-free accurate separation of the targeted cells using procedures that do not require a bio-chemical laboratory. Thus, the separation method proposed below is based on dielectrophoresis (DEP) phenomenon that has been proved to yield very good results in separating targeted groups of cells of known properties5. However, the present work goes one step forward toward cell-type identification based on the specific signature when imaged through a radio frequency (RF) signal. This approach sets another check point in the identification of living cells which makes the method more accurate than the DEP separation. If two cells are biologically different but they encounter same size and complex dielectric constant, they will be identified by the RF imaging. Hence, the second step in the process has a double-fold objective: to accurately identify among the separated cells in the first phase and to attempt to identify them when a comprehensive database will be developed. The extra step ensures the accuracy of the separation process and further enables identification of the source of the unlabeled individual circulation tumor cells (CTCs) based on their specific signature as a response to a GHz range RF weak signal. The advantage of the principle is found in capability of automation and portability of the platform. Hence, two different platforms are used in this work. One is built to separate CTCs based on their size and complex dielectric permittivity using the DEP process. The second is built to identify the response signature of the already separated cells to RF signal with the objective of identification of the type of cell. Below, a brief state of the art of living cell separation and identification is presented.
Separation of rare cells from treated blood or media has been performed for quite some time to the extent that such methods are currently used in clinical tests. Flow cytometry enables counting and separation of specific cells from a sample of mixed cells based on their affinity to a selected die or a labeling agent. For an example, a mix of acid fuchsin, acridine yellow, and berberine was used by Friedman6 for uterine cancer detection. Since then, multiple recipes used for detection of cells of interest as selected matching antibodies have been made available. Flow cytometry counting as well as separation are both possible and some of such found cells may be re-imaged in cultures under certain circumstances7. The flow cytometry has evolved to the extent that markers could be designed for the specific molecules that differentiate the functionality of same type of cells or it uses novel detection principles such as optical impedance8,9.
Separation of rare cells has also been successfully carried out using methods based on the antibody-antigen affinity. Thus, magnetic microbeads coated with the antibody of interest are mixed with treated blood. Cells that overexpress the targeted antigens will collect such magnetic particles based on their bio-chemical expression. Further, the mix is inserted in a magnetic field which will attract the magnetic particles and the cells attached to them while the unattached cells will be flushed with the flow stream. The above procedure was employed in developing equipment and in clinical protocols that have been intensively used10,11.
One of the basic principles that aided to set the objectives of this research was focused on the ability of the method to adjust to the variance of the searched cells related to size as well as their expression. If most of the CTCs size ranges usually between 10 μm and 25 μm, two cells of the same origin and expression may have different sizes. If separation is exclusively performed based on size, such variance may yield to the reduction of the effectiveness of the separation method. DEP will perceive size just in the amplitude of the attraction or rejection force exerted on the cells and not in the type of force. The type of force is given by the complex dielectric constant that will be similar for all cells of same type and which will give the cross-over frequency value. This is the main rationale that leads this work toward a separation principle that enables variance as well as applies multiple criteria of selection such that if a cell fails one of the criteria, there are chances to be identified by the others. The clinically accepted cell count method10, CellSearch, uses the amount of 7.5 cc of fresh blood that carries about 40 × 109 cells. Out of that count, any number below 102 CTCs is found in a fresh blood specimen.
The interest toward developing microfluidic devices is primarily driven by the high ratio of surface area to volume which enhanced the ability to probe a unit volume into a detailed discrete biological sample. In addition, microfluidic platforms for cell separation and identification enjoy all the advantages associated with the microfluidic devices. These include increased functional integration, reduced volume of chemicals (samples and reagents) and power consumption, enhanced transport phenomena leading to reduced time to results, and improved portability due to miniaturization12,13. Microfluidic devices have been intensively investigated, developed, and characterized for improved detection and separation of particular cells from whole blood or other fluid bio-specimens12–14. Different principles of separation and counting under still and continuous flow conditions have been successfully developed and largely used by preparing immuno-coated surfaces that attract and retain the targeted cells. Similarly, antibody conditioned magnetic microbeads have been used to uphold the targeted cells under magnetic field in the continuous flow microfluidic circuit. This principle was extended into a very successful product that is used in evaluation of the effectiveness of chemotherapy15. The separation based on magnetic microparticles method has, however, a long history despite the fact that there are few drawbacks of the method16,17. These evidences provide good rationale toward the support of the ongoing investigations on other suitable technologies of accurate separation and counting of rare cells. Accuracy of any method could be improved by using multiple checking rules. For an example, the isolation by size of epithelial tumor cells is based on size only and cells smaller that the selected threshold may fall in the same category16. In cytometry as in magnetic microparticle separation, cells are identified based on specific cell overexpression of antibodies on which the label attaches. Clustering of cells may represent a significant error-inducing factor of both methods. Microsystems devices bring another approach in cell separation and identification. Cells could be separated in bulk, in large microfluidic chips, or they could run as single cells through functionalized microchannels. The advantage of the controlled flow enables the use for identification of cells which would be assessed based on their specific image perceived by the suitable selected detection physics. Recently, several microfluidic platforms were reported18 for detection, and separation of cells have been reported. Certain microfluidic devices use biomarkers that are selected for the specific cells to separate and differentiate13,16,17,19–21. The commonly utilized biomarker of CTCs is epithelial cell adhesion molecule (EpCAM; antigen). It is widely accepted that the EpCAM is overexpressed on the surface of most CTCs released from carcinomas. Based on this characteristic of CTCs, microfluidic devices have been developed for separation of CTCs from blood based on the concept of antigen-antibody affinity22. The surface of such devices is coated with anti-EpCAM (antibody) to which CTCs from blood adhere and are later collected for analysis. As an example of another bio-label utilized for capture of prostate cancer CTCs, in same manner as above described is prostate-specific membrane antigen (PSMA). PSMA is usually over-expressed by prostate cancer cells and the corresponding anti-PSMA antibody is known as J59123. This separation approach is beneficial as it acts independent of CTCs diameter, thereby preventing other cells such as leukocytes from being wrongfully identified and separated as CTCs since the diameter range of leukocytes is similar with that of CTCs. Hence, an antigen-antibody combination has to be available for the searched CTCs to implement this technique and this may not always be the case. All the above methods show remarkable benefits as well as few drawbacks. Thus, different principles of separation are investigated; each yielding certain benefits over others19. The above reasoning leads toward new principles of effective separation and accurate identification. One such separation principle, the identification method, the test procedure, and the results are presented below.
Materials and methods
In this work, DEP is used as the separation principle. DEP refers to the movement of electrically neutral, but polarizable particles, i.e. cells in the case of CTC separation, in a non-uniform electric field. When the cell moves toward the gradient of electric field maxima and gradient of electric field minima, then the DEP phenomenon is specifically termed as positive-DEP (pDEP) and negative-DEP (nDEP), respectively. The force that positions a cell in a non-uniform electric field is determined by several factors including cell diameter, electrical conductivity of the medium, and capacitance of the cell membrane and frequency of the external electrical signal. The frequency may play the role of a moderator in capturing cells with size variations. Judicious selection of nDEP and pDEP improves the selectivity of the method and the capability of targeted cells of being retained for separation. Therefore, for a specific cell diameter, electrical conductivity of the medium and frequency of the external electrical signal on the electrode toward which the cell moves are determined solely by the capacitance of the cell membrane. As capacitance of the cell membrane varies between cell types, it is possible to have at a given frequency range both pDEP and nDEP within the same blood specimen, thereby enabling the separation of particular cells from the rest of other cell types in the blood sample. Polarizable spherical cell suspended in low-conductivity medium and placed under non-uniform electrical field experiences a DEP force that is given by24:
where ε0, εm are the vacuum and suspending medium relative dielectric constant, respectively, r is the radius of the cell, is the real part of Clausius–Mossotti factor, and E represents the peak electric field. Clausius–Mossotti factor fcm has real and imaginary parts. The real part of fcm determines DEP on cell and imaginary part of it describes the electrorotational torque on the same. Clausius–Mossotti factor is defined as25:
where and are the frequency dependent complex permittivity (CP) of the cell and of the suspending medium as function of the angular frequency ω, respectively. The practical design takes advantage of the above phenomenon. Thus, cells that are searched by both size and constant dielectric value will be able to travel through the electrodes, while all the other cells will be retained by the electric field created by the electrode. The cut-off frequencies and the preparation of blood specimens are given in detail in our previous work24,26. Figure 1 illustrates four snapshots taken to cells retained by the nDEP, while the marked MDA231 cancer cell is seen to travel the electrode and settle in high electric filed area. Selection of the MDA 231 cell line model was suggested by the extended data availability on this type of cell line.

Illustration of the sequence of a separation process for MDA231 cells (circled) from blood specimen. 20 V peak-to-peak and frequency of 30 kHz, medium conductivity is 100 µS cm−1, and electrodes width is 15 µm.
Cells of interest are targeted by the suitable selected frequencies20,26,27. The cells seen in the lower left part of the figure will not cross the comb-like electrode and will be directed to flow to the selected outlet. The targeted cells will be allowed to cross the electrodes due to the weak pDEP force they experience and will be pushed to flow to a different outlet where they will be collected. The images are shown in sequence from top-left to bottom-right.
Fresh blood samples were suspended in the low-conductivity sucrose/dextrose medium with ratio of 1:10. A small amount of trypsin is added to the sucrose–dextrose medium to bring the conductivity of the mix to 100 µS cm−1 24,26,28. The two types of cells are mixed and the separation process is performed through the chip as shown in Figure 2. Cells through and separated are counted after the separation using a hemocytometer. The group of tests was carried out while focusing on high ratio between blood cells and CTC count. Diluted blood cells were prepared as above. The cancer cell lines were diluted in 1:14 and 1:105 volumes and fed through the separator. After few minutes of separation, the supply of cells was cut. The separated cells were counted under the microscope and the accuracy of separation was calculated. The separation was performed again and the cells were counted each time. The results show that the separation rate is superior to 95%26 in both circumstances as shown in Table 1. For large number of cells in solution, counting is hardly relevant as the counting number ends in four zeros. However, it was noticed that the counting was consistent for each experiment and given the fact that the direct counting for reduced number of cells show separation efficiency above 95%, and following reference5, we concluded that the efficiency of separation for rare cells in solution is exceeding 95%. Each experiment was repeated in excess of 50 times.

(a) The configuration of the electrodes—design layout; the distance between the long electrodes is 30 µm and the digits-like electrodes are 15 µm side squares. (b) The separation chip assembled is shown with a Canadian dollar coin (Ø 26.4 mm) in the picture.
The chip was designed to perform separation of 10 cc of diluted cells in media in about 40 min. The width of the chip is of 100 mm and the gap between the aligned electrodes on the top and bottom glass is of 200 µm. The separation was performed at average flow velocities below 250 µ ms−1. The fluid velocity was controlled using a syringe pump. Higher flows may lead to loss of targeted cells. The two sets of electrodes on the top and bottom glass were carefully aligned before the chip was sealed. Holes drilled in the glass enable smooth flow of the mix. The electrodes are fed through the board connector that enables fast change in the electrical settings on the individual electrodes. The thin metallic electrodes (about 0.2 µm thick) were deposited on glass using conventional micromachining.
Further to separation, the rare cells have been subjected to the identification process. This procedure was carried out for a few types of cell lines. Selection of the imaging frequency is dependent on the Claussius–Mossotti factor expressed above. In general, a material can be characterized by its unique permittivity signature and conductivity or CP and these parameters are mainly determined by the molecular structure of the material24. In fact, the dielectric properties of a material hint on how that material will interact with an applied electromagnetic field. The electromagnetic measurement spectrum of the CP covers from low frequencies to much higher microwave frequencies. Once these properties are known, the physical and chemical properties of the material can be estimated. Permittivity and conductivity information at low frequency are used in separating and transporting cells by DEP. Higher frequency CP measurements were performed for cell characterization required for identification and detection. Relative or normalized CP is written as:
where is the real part of the CP or the dielectric constant and is the imaginary part of the CP, σ is defined as the conductivity including both the material conductivity and the dielectric loss, ω is the angular frequency of the applied electric signal, and .
Detection of single cell at radio-frequencies requires the characterization of each cell dielectric properties for identification. Unfortunately, single cell measurement is impossible and measurements are made on tissues and cells in bulk29. Hence, CP of cells in bulk form suspended in media is measured with minimum amount of media using resonant cavity perturbation method30. Very small sample volume is used with this method with appropriate and accurate measurement results. From the effective CP (εeff) measurement of cells in the media (εm) and the volume fraction of particles (Vf), single cell or particle CP (εp) is estimated by the Maxwell–Wagner–Hanai formula31 given by:
For separation as mentioned above, cells are suspended in a media with a mixture of water and sucrose/dextrose20,26,27. For cell detection, since the CP of the solution of sucrose/dextrose is very close to that of the cells, a mixture of glycerol with trypsin (water soluble triglyceride) is used in experiments due to its very low dielectric constant at RF frequencies. Trypsin is mainly used to avoid cells clustering and to condition the dielectric constant of the media much different from the one of the cells. The duration of the essay is of 15–20 s which represents the travel of the cell through the interrogation station. However, cells are moved in glycerol–trypsin environment prior to the tests. In a single platform, duration of switch from sucrose–dextrose media to glycerol–trypsin may be as long as 2 min. However, cells survive in glycerol–trypsin environment for up to few days but the cell proliferation is inhibited32.
The medium preparation and the conductivity measurements for separation are found in Gleghorn et al. and Alazzam et al.23,26. For cells identification, a mixture of glycerol–trypsin solution 0.1 mol in a ratio of 3.5:1 yields dielectric constant of 18 and dielectric loss of 18 in the RF measurement band. This choice of media solution improves the sensitivity of the measurement and it does not produce harm to the cells as glycerol is used in bacteria culturing30. During the entire measurement, cells are kept in the appropriate media for their survivability and health.
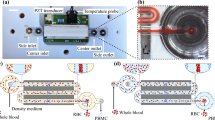
The measurement platform is realized in same technology steps as the separation chip. More attention is given to the connections used for high frequency injection and reading to avoid any inaccurate interpretation. The configuration of the chip and a picture of the platform are given in Figure 3.

(a) and (b) The interrogation platform—detail of the microfluidic channel, the focusing electrodes, and the detection/imaging electrodes. Part (a) illustrates the inlet and the outlet and part (b) illustrates the detail of the microfluidic circuit. (c) The cell identification and imaging platform. The microfluidic chip is covered by an aluminum case. The visibility of the microfluidic channel is ensured by a visitation access cutout.
The width of the electrode was selected as 50 µm. The channel is 50 µm in height and 300 µm in width. The emitter and the antenna (reversible configuration) require an accurate alignment as they are positioned on two different microfluidic surfaces. The measurement channel includes four focusing electrodes that are activated to the frequency that initiates nDEP. This enables the cells to align and center within the channel while pressure is driven to the interrogation post. The two transversal electrodes use impedance measurement to detect the existence of the cell approaching the interrogation point. Once a cell is detected, an interrogation signal with frequencies ranging from 1 to 12 GHz is emitted at the time the cell reaches the interrogation point. The intensity of the injected signal is of 1 mW and the duration of the sweep signal is of 100 ms. The flow average velocity was set to 10–20 µm s−1.
Results
The main contribution of this paper is on using multiple criteria of separation for accurate identification of cells. The DEP part was presented25, such that the results of concern are mainly the ones obtained from the RF response collected from individual cells.
The results of the RF identification measurements carried out on several cancer cell lines. The measurements of the CP for the two cell lines are shown in Figure 4.

(a) The real part of the CP for 92.1 and white blood cell (WBC) cells. (b) The imaginary part of the CP for 92.1 and WBC cells. Tests were repeated six times. The figure shows the variation of the CP values.
Both real and imaginary parts of the CP are shown for imaging frequencies between 2 GHz and 4.5 GHz. A certain intra-cellular variance of both real and imaginary parts is observed. Since CP of particles at high frequency shows dispersion with frequency, measurement range should be selected such that the dielectric constant and the dielectric loss are non-dispersive28. From the measurement of the CP of a few cancer cells lines, the frequency range 2–4.5 GHz shows very small dispersion and therefore this frequency range is selected for cell identification by RF imaging.
A sample of the insertion loss measurement using the vector network analyzer type 8720ES is illustrated in Figure 5. The lines indicate the cumulated average measurement for the cells that were investigated.

Results of the imaging. Here the range of measurement is extended from 1 GHz to 12 GHz. The plots indicate average measurements for (a) top—insertion loss and (b) bottom—return loss. The three lines indicate the insertion loss expressed in dB for the media (dashed line—glycerol and water in proportion of 3.5:1 and trypsin to achieve the conductivity of 150 S m−1) (dashed line—media, blue line—uveal melanoma cells type 92.1 in media as above, black line—Jurkat WBCs in media as above).
The measurements revealed that cells of same origin yield identical RF insertion loss. The selection of glycerol based media helps improving the sensitivity of the measurement as the cell RF properties are detected and not the media. From identification perspective, the presented RF imaging method is extremely suitable to differentiate between two different types of cells. To support this statement, in Figure 6, the insertion losses for two types of cells are illustrated within the range of 2–4.5 GHz. The repeated measurements for the same type of cells show no abatement from the plots shown in Figure 6. The explanation for this consistency is due to the selected measurement scale—in dB which in terms of signature is consistent for the same line of cells.

Results recorded on the RF insertion and RF return losses for two types of cells: uveal melanoma line 92.1 and WBC (Jurkat leukemia WBCs). (a–c) show the insertion loss signal between 2 GHz and 4.5 GHz for (a)—WBC from which media is extracted, (b) UM 92.1 from which media was extracted, and (c) the difference of the two signals in (a) and (b). (d–f) show corresponding results for the return losses: (d) WBC from which media response was extracted, (e) UM 92.1 from which media was extracted, and (f) the difference of the two signals.
The results show that the signature of each type of cell is significantly different, while the variation within the same type of cell is minimal—virtually undetectable with the used setup. However, the identification will require the development of an extensive atlas of signatures that could be used of automatic recognition from a database. Further, separation of the cells will pose no challenge as long as cells are identified. The method yields 100% accuracy based on the signature of the RF response of the cells to low intensity large spectrum RF signal.
Discussion
The above presented results reveal the capability of the discussed method of identification based on RF imaging to be extended toward cell type identification. Hence, the presented results do point out clearly that the RF imaging represents a very good label-free tool for cell identification after separation and the capability of the method shows great potential for cell typing. The two platforms that complete the label-free separation and the identification use same technology. They could be integrated in a unique platform that would perform fully automatic both separation and identification. Besides the insertion loss, one could extract the return loss as well and the detailing of the two yields more hints and better objective identification. Both the insertion loss and the return loss could be used for identification and validation. It is expected that the results will be dependent on the specific configuration of the interrogation station which will modify the configuration of the microchannel. However, the amplitude of the RF signal may vary in a different configuration of the interrogation station, but the signature is expected to reveal the type of cell. The development of the database of type of cells is a feasible task as the platform may be easily automatized. The integration of the separation and identification platforms in a sole piece and extension of the capability of operation of the separation in parallel configuration could lead to time reduction of the process making it a great candidate for a point of care device. The proposed platform has the advantage that no calibration equipment is needed as the reference signal could be taken from the media used here as a baseline. Few of the advantages of such a platform are: portability, automation, capability to perform accurate assessment with little volume specimen—single living cells. The principle of detection could be used for evaluation of cells that may be different from their predecessor: infected cells, mutant cells, etc. The two-steps principle is expected to yield efficiencies higher than the ones reported in the literature1,5,16 as the first criterion of separation may be relaxed such that all targeted cells would pass it. The second step RF signature identification will collect only the cells of interest based on their specific signature.
Besides counting, a suitable database of the signature of the cancer cells could enable fast and accurate detection of the source of a CTC found in the blood stream. This may significantly improve the output of the treatment as long as the type of cancer in the metastasis is known.
References
Editorial. The future of cancer genomics. Nature Medicine 2015; 21: 99–99.
Feero WG, Guttmacher AE, McDermott U et al. Genomics and the continuum of cancer care. The New England Journal of Medicine 2011; 364: 340–350.
Allard WJ, Matera J, Miller MC et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clinical Cancer Research 2004; 10: 6897–6904.
Nagrath S, Sequist LV, Maheswaran S et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007; 450: 1235–1239.
Gascoyne PR, Wang X-B, Huang Y et al. Dielectrophoretic separation of cancer cells from blood. IEEE Transactions on Industry Applications 1997; 33: 670–678.
Friedman H. The use of ultraviolet light and fluorescent dyes in the detection of uterine cancer by vaginal smear. American Journal of Obstetrics and Gynecology 1950; 59: 852–859.
Chirumbolo S, Vella A, Ortolani R et al. Differential response of human basophil activation markers: A multi-parameter flow cytometry approach. Clinical and Molecular Allergy 2008; 6: 12.
Cheung K, Gawad S, Renaud P. Impedance spectroscopy flow cytometry: On-chip label-free cell differentiation. Cytometry Part A: Journal of International Society for Analytical Cytology 2005; 65: 124–132.
Díaz-Ramos MC, Engel P, Bastos R. Towards a comprehensive human cell-surface immunome database. Immunology Letters 2011; 134: 183–187.
Farace F, Massard C, Vimond N et al. A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas. British Journal of Cancer 2011; 105: 847–853.
Miltenyi S, Müller W, Weichel W et al. High gradient magnetic cell separation with MACS. Cytometry 1990; 11: 231–238.
Autebert J, Coudert B, Bidard F-C et al. Microfluidic: An innovative tool for efficient cell sorting. Methods 2012; 57: 297–307.
Chen J, Li J, Sun Y. Microfluidic approaches for cancer cell detection, characterization, and separation. Lab on a Chip 2012; 12: 1753–1767.
Arya S, Lim B, Rahman ARA. Enrichment, detection and clinical significance of circulating tumor cells. Lab on a Chip 2013; 13: 1995–2027.
Muller V, Riethdorf S, Rack B et al. Prognostic impact of circulating tumor cells assessed with the CellSearch System and AdnaTest Breast in metastatic breast cancer patients: The DETECT study. Breast Cancer Research 2012; 14: R118.
Vona G, Sabile A, Louha M et al. Isolation by size of epithelial tumor cells: A new method for the immunomorphological and molecular characterization of circulating tumor cells. The American Journal of Pathology 2000; 156: 57–63.
Xu H, Aguilar ZP, Yang L et al. Antibody conjugated magnetic iron oxide nanoparticles for cancer cell separation in fresh whole blood. Biomaterials 2011; 32: 9758–9765.
Didar TF, Li K, Tabrizian M et al. High throughput multilayer microfluidic particle separation platform using embedded thermoplastic-based micropumping. Lab on a Chip 2013; 13: 2615–2622.
Esmaeilsabzali H, Beischlag TV, Cox ME et al. Detection and isolation of circulating tumor cells: Principles and methods. Biotechnology Advances 2013; 31: 1063–1084.
Osman O, Toru S, Dumas-Bouchiat F et al. Microfluidic immunomagnetic cell separation using integrated permanent micromagnets. Biomicrofluidics 2013; 7: 054115.
Tsutsui H, Ho C-M. Cell separation by non-inertial force fields in microfluidic systems. Mechanics Research Communications 2009; 36: 92–103.
Baeuerle P, Gires O. EpCAM (CD326) finding its role in cancer. British Journal of Cancer 2007; 96: 417–423.
Gleghorn JP, Pratt ED, Denning D et al. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab on a Chip 2010; 10: 27–29.
Alazzam A, Roman D, Nerguizian V et al. Analytical formulation of electric field and dielectrophoretic force for moving dielectrophoresis using Fourier series. Microfluidics and Nanofluidics 2010; 9: 1115–1124.
Lapizco-Encinas BH, Rito-Palomares M. Dielectrophoresis for the manipulation of nanobioparticles. Electrophoresis 2007; 28: 4521–4538.
Alazzam A, Stiharu I, Bhat R et al. Interdigitated comb-like electrodes for continuous separation of malignant cells from blood using dielectrophoresis. Electrophoresis 2011; 32: 1327–1336.
Puttaswamy SV, Sivashankar S, Chen R-J et al. Enhanced cell viability and cell adhesion using low conductivity medium for negative dielectrophoretic cell patterning. Biotechnology Journal 2010; 5: 1005–1015.
Braschler T, Demierre N, Nascimento E et al. Continuous separation of cells by balanced dielectrophoretic forces at multiple frequencies. Lab on a Chip 2008; 8: 280–286.
Markx GH, Davey CL. The dielectric properties of biological cells at radiofrequencies: Applications in biotechnology. Enzyme and Microbial Technology 1999; 25: 161–171.
Hakki B, Coleman P. A dielectric resonator method of measuring inductive capacities in the millimeter range. Microwave Theory and Techniques, IRE Transactions on 1960; 8: 402–410.
Bordi F, Cametti C, Gili T. Dielectric spectroscopy of erythrocyte cell suspensions. A comparison between Looyenga and Maxwell–Wagner–Hanai effective medium theory formulations. Journal of Non-crystalline Solids 2002; 305: 278–284.
Wiebe J, Dinsdale C. Inhibition of cell proliferation by glycerol. Life Sciences 1991; 48: 1511–1517.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0
About this article

Cite this article
Stiharu, I., Alazzam, A., Nerguizian, V. et al. Single living cell manipulation and identification using microsystems technologies. Microsyst Nanoeng 1, 15031 (2015). https://doi.org/10.1038/micronano.2015.31
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/micronano.2015.31
This article is cited by
-
Transparent, patterned graphene oxide films with tunable electrical conductivity using thermal, chemical, and photoreduction techniques for lab-on-a-chip applications
Analytical and Bioanalytical Chemistry (2023)
-
A Handheld Electronics Module for Dielectrophoretic Impedance Measurement of Cancerous Cells in the Microchip
BioChip Journal (2018)
