
Abstract
When apoptosis is initiated by manganese (II) loading, hyperthermia or thapsigargin treatment, human HL-60 and AsPC-1 cells initiate de novo synthesis of the C5a receptor (C5aR) and generation of its ligand, the ribosomal protein S19 (RP S19) homodimer. The ligand–receptor interaction, in an autocrine/paracrine fashion, promotes apoptosis, which can be bypassed by exogenous administration of C5a, another ligand. The proapoptotic function of the RP S19 dimer is reproduced by a C5a/RPS19 chimera that contains the body of C5a and the C-terminal region (Ile134-His145) of RP S19. The RP S19 dimer or C5a/RPS19 and C5a inversely regulate the expression of Regulator of G protein Signaling 3 (RGS3) gene in the apoptosis-initiated cells. Namely, the RP S19-type proteins upregulate RGS3 expression, whereas the C5a reduce it. Transformation of HL-60 cells to overexpress RGS3 promotes apoptosis in association with the downregulation of the Extracellular signal-Regulated Kinase (ERK) signal, and vice versa in the RGS3 knocked-down cells. Consistent with this result, an inhibitor of ERK phosphorylation effectively enhances the apoptotic rate in wild-type HL-60 cells. Moreover, a dominant negative effect on the RP S19 dimer production encourages apoptosis-initiated HL-60 cells with a longer lifespan in mouse than the natural effect. Our data indicate that, in apoptosis-initiated cells, the ligand-dependent C5aR-mediated dual signal affects the fate of cells, either apoptosis execution or survival, through regulation of RGS3 gene expression and subsequent modulation of ERK signal.
Similar content being viewed by others


Main
The major monocyte-specific chemoattractant in rheumatoid arthritis-synovial1 and atherosclerosis-aortic2 lesions has been identified to be the ribosomal protein S19 (RP S19) homodimer. The RP S19 dimer has an intermolecular cross-linkage between Lys122 and Gln137 generated by a transglutaminase-catalyzed reaction,3 and it attracts not only human monocytes, but also guinea pig and rabbit monocytes4, 5 on binding to the C5a receptor (C5aR).6 C5aR is also present on neutrophils even more abundantly. In striking contrast, the RP S19 dimer does not attract neutrophils and even antagonizes the interaction between C5a and C5aR.6 We earlier identified the C-terminal moiety of RP S19 (Ile134-His145) as the switch converting it from the agonist to the antagonist for the C5aR on neutrophils.7, 8 The RP S19 dimer is commonly released from apoptotic cells such as HL-60 cells, a leukemia human cell line,9 AsPC-1 cells, an epithelial human cell line,10 and NIH3T3 cells, a fibroblastoma-derived mouse cell line.11 In vivo experiments show that apoptotic HL-60 cells injected into the rabbit skin were rapidly cleared by infiltrating monocytes/macrophages in response to the RP S19 dimer liberated from the apoptotic cells. These macrophages, digesting engulfed apoptotic cells, moved to the regional lymph nodes through lymph vessels and induced an acquired immune response by means of the presentation of the apoptotic cell-derived antigens to T cells.5 During these phenomena, signs of acute inflammation including neutrophil infiltration were not observed. These findings led us to propose that the RP S19 dimer and the C5aR of monocytes play roles in phagocytic clearance and immunological monitoring of apoptotic cells without phlogistic stimulation.12
We recently showed another phenomenon mediated by the RP S19 dimer and C5aR in apoptosis (the RP S19 dimer-C5aR system).11 On release from manganese (II)-induced apoptotic NIH3T3 cells, the RP S19 dimer accelerated apoptosis through the C5aR, although these cells had not been thought to constantly express C5aR. It is surprising that the C5aR was de novo synthesized by this cell type after the apoptotic initiation. Consistent with this finding, exogenous administration of C5a to the apoptosis-initiated NIH3T3 cells reversed the initiation of apoptosis and led to survival, as observed earlier in neutrophils (the C5a-C5aR system).11, 13 Namely, C5aR has a ligand-dependent pro- and anti-apoptotic dual function, and the dual function regulates, at least in part, the life span of apoptosis-initiated cells.
The apoptotic process is sometimes divided into three phases, namely, the initiation, the effector, and the execution phases.14, 15 Our proposal is that the apoptosis-initiated cells integrate endogenous and microenvironmental information and make the decision to die or to live; the C5aR de novo synthesis and the RP S19 dimer generation would be active in the effector phase and participate in the information collection.
Despite this model, the question still remains as to why a leukocyte chemotactic receptor plays such an important role in apoptosis. A literature search on other leukocyte chemotactic receptors in terms of apoptotic regulation found reports on apoptosis promotion by annexin-1 (ANXA1) through Formyl Peptide Receptor (FPR) and FPR like 1 (FPRL1) in neutrophil apoptosis.16 FPR and FPRL1 are members of the G protein-coupled receptor (GPCR) family, as in the case of C5aR. ANXA1 was released not only from neutrophils, but also from dexamethasone-treated macrophages17 and from anti-Fas IgM-treated Jurkat T cells in a caspase-dependent manner.18 On the other hand, ANAX1 was reported to be a receptor antagonist of FPR and FPRL1 in neutrophil chemotaxis induced by formyl-Met-Leu-Phe (fMLP).19, 20, 21 Collectively, ANXA1 antagonizes neutrophil chemotaxis in a short period of time, and it promotes apoptosis in a long period of time,22 as does the RP S19 dimer described above. One could ask whether the RP S19 dimer accelerates neutrophil apoptosis through C5aR and whether FPR and/or FPRL1 is de novo synthesized in apoptosis-initiated cells other than leukocytes and whether ANXA1 accelerates apoptosis through the receptors.
Before pursuing this line of investigation, the question of whether or not the pro-apoptotic effect of the RP S19 dimer-C5aR system, shown in manganese (II)-loaded NIH3T3 cells, is common in human cell lines under various apoptotic initiations needs to be addressed. To confirm the pro-apoptotic effect of the RP S19 dimer, we also need to address whether a chimeric protein that bears the body of C5a and the C-terminal region (Ile134-His145) of RP S19 (C5a/RPS19) reproduces the pro-apoptotic effect.23 As preparation processes of the cross-linked homodimer of RP S19 are very complicated in vitro,3, 10 we currently prepared C5a/RPS19 as a substitute for the RP S19 dimer and at least showed reproduction of the C5aR-mediated antagonism to neutrophils in chemotaxis.24 In this report, we describe the occurrence of the de novo synthesis of C5aR and apoptosis promotion by the RP S19 dimer or C5a/RPS19 through C5aR in human HL-60 and AsPC-1 cell lines under three kinds of apoptotic initiations: hyperthermia,9, 10 manganese (II) loading,11, 25 and thapsigargin treatment.26
We then addressed the question of whether the proapoptotic effect of the RP S19 dimer was caused by C5aR antagonism or by C5aR agonism. Earlier, the former idea was supported, as the common view was that GPCR molecules would alternate between an inactive and active conformation, and, therefore, a proportion of the GPCR molecules could generate intracellular signals even in the absence of a ligand agonist.27 The apparent constitutive activity of the GPCR might generate the survival signal.28 For this reason, the proapoptotic effect of RP S19 might result from the antagonism to the C5aR-derived survival signal. However, it is also possible that the RP S19 dimer actively blocks the GPCR-derived survival signal. Therefore, we examined the upregulation of any regulator of G protein signaling (RGS) on the ligation of C5aR by the RP S19 dimer or C5a/RPS19. We found that RGS3 was upregulated by de novo synthesis and strongly promoted apoptosis. Our results indicate that the RP S19 dimer behaves as an agonist in the C5aR-mediated apoptosis promotion.
MATERIALS AND METHODS
Animals
Kud:ddY and CAnN.Cg Foxn1nu/CrlCrlj (nu/nu) strain female mice (15–20 g body weight), which were specifically pathogen free, were purchased from Kyudo Corp. (Kumamoto, Japan). They were maintained in the Center for Animal Resources and Development, Kumamoto University. The animal experiments were performed in three different experiments with duplicates under the control of the Ethical Committee for Animal Experiment, Kumamoto University School of Medicine. The mice were killed under ether anesthesia.
Materials
The inhibitors, antibodies, plasmid vectors, and primers used in the current study are listed in Supplementary Table S1–S3. The methods used for preparation of the mutant HL-60 cells, the knocked down HL-60 cells, and their control mock HL-60 cells are described in the legends of Tables.
Cell Cycle Analysis
DNA content was analyzed using a FACS Caliber flow cytometer (BD, Tokyo, Japan) according to a procedure established by Papazisis et al29 In brief, aliquots of cells were harvested at several time points after initiating apoptosis and fixed in 70% ethanol at −20°C for 3 h. The cells were stained with 20 μg/ml propidium iodide (PI) containing 20 μg/ml RNase A for 30 min. The cell populations of the G1 subset or at the G0/1, S, and G2/M phases were quantitatively determined using Multicycle Cell Cycle Software. The percentage of G1 subset cells of the total number of cells was used as the apoptotic ratio. For the percent inhibition of apoptosis induced by a particular reagent, we calculated this at 24 h by: ((control apoptotic ratio at 24 h)−(experiment apoptotic ratio at 24 h))/(control apoptotic ratio at 24 h) × 100.
Fluorescence-Activated Cell Sorting (FACS) Analyses
For analysis of cell surface expression of C5aR and phosphatidylserine exposure, cells were suspended in PBS containing 5% fetal bovine serum (FBS) and 5 mM CaCl2 (FACS medium). Cells were incubated with fluorescein isothiocyanate (FITC)-conjugated anti-C5aR monoclonal antibody and phycoerythrin (PE)-conjugated annexin V for 30 min at 4°C.
For analysis of cytoplasmic RGS3, cells were pretreated in FACS medium with 0.01% saponin for 15 min at 4°C to induce cell membrane pore formation and incubated with normal rabbit IgG for 20 min at 4°C to block non-specific IgG binding. After cells were incubated with anti-RGS3 rabbit IgG for 30 min at 4°C, PE-conjugated anti-rabbit goat IgG was reacted for another 30 min. Cell nuclei were sometimes stained with PI.
For analysis of number of alive human cells in mice, HL-60 cells (107 cells) were labeled with Red or Green Fluorescent SYTO* Nucleic Acid Stains, SYTO*59, or SYTO*14 (Invitrogen, California, USA), for 1 h at 22°C according to the manufacturer's instruction manual. After washing in PBS twice, the HL-60 cells were treated in 50 μl of PBS containing 0.5 mM MnCl2 for 3 h at 22°C and injected into peritoneal cavities. After 24 h, the HL-60 cells were recovered with 10 ml of cold PBS and were re-suspended in FACS medium to examine the cell surface expression of phosphatidylserine exposure. Cells were incubated with FITC-conjugated or PE-conjugated annexin V for 30 min at 4°C.
Fluorescent Immunocytochemistry
For analyses of cell surface expression of C5aR and phosphatidylserine exposure, cells were stained with FITC-conjugated anti-C5aR monoclonal antibody and PE-conjugated annexin V for 40 min at 4°C in FACS medium. For analysis of cytoplasmic RGS3, cells were washed twice in PBS and fixed in 1% (w/v) paraformaldehyde for 1 min, then they were pretreated in FACS medium with 0.5% Triton X-100 for 15 min at 4°C to induce cell membrane pore formation. After incubating with normal rabbit IgG for 20 min at 4°C to block non-specific IgG binding, anti-RGS3 rabbit IgG was reacted for 40 min at 4°C and FITC-conjugated anti-rabbit goat IgG was reacted for another 30 min at 4°C. Cell nuclei were stained with PI. Cells were observed using an Olympus Fluoroview FV300 microscope (Olympus, Tokyo, Japan).
Chemotaxis Assay
Monocytes and neutrophils were isolated from heparinized human venous blood of healthy donors as described earlier.30 They were suspended in RPMI1640 containing 10% FBS and HBSS containing 0.5% BSA for the multiwell chamber assay. The multiwell chamber assay was carried out according to the method of Falk et al,31 using a Nucleopore filter with a pore size of 5 μm (Bethesda, MD, USA) for monocytes and 3 μm for neutrophils. After incubation for 90 min, each membrane was separated, fixed with methanol, and stained with Giemsa solution. The total number of migrated cells beyond the lower surface of the membrane was counted in five microscopic high-power fields. The results are expressed as the number of migrated cells.
Calcium Imaging
Neutrophil-like phenotypes differentiated from HL-60 transformants were loaded with the calcium-sensitive Fura 2-AM (1 μM) in HBSS containing 20 mM HEPES and 3% BSA (pH 7.4) for 30 min at 37°C according to manufacturer's instructions (Dojin Chemical Institute Co., Kumamoto, Japan). After washing with buffer, samples were placed directly into the cell suspension in a cuvette after a 5-min baseline recording. Recordings were made with an F-2500 calcium imaging system with FL Solutions (HITACHI, Tokyo, Japan) that calculated the ratio of fluorescent signals obtained at excitation wavelengths of 340 and 380 nm at 37°C.
Respiratory Burst Reaction
Superoxide anion production by cells’ NADPH oxidase during the respiratory burst reaction was determined using WST-1 as described earlier.23 Reactions were measured in a HITACHI U-2000A spectrophotometer (HITACHI) using a 1-ml cuvette with a reaction mixture containing the cell suspension, WST-1 (500 μM), and cytochalasin B (5 μM). The cuvette was kept at 37°C, the stimulus was added, and absorbance of the reduced formazan was recorded continuously at 438 nm.
SDS-PAGE and Western Blot
Electrophoresis was performed using 10 and 15% polyacrylamide gels according to the method of Laemmli.32 Culture supernatants or cell pellets were collected at several time points after apoptotic stimulations. Proteins in supernatants were immediately concentrated 1000-fold using 5 KDa cut-off Amicon Ultra® concentrators (Millipore Corporation, Bedford, MD USA). Proteins in cells were extracted in loading buffer with 1% SDS, Triton-X100, and CHAPS. Samples were applied to SDS-PAGE and transferred to an Immobilon-PSQ membrane. After treating with 1% Block Ace™ (Dainihonseiyaku, Osaka, Japan) for 1 h at 22°C, the first rabbit IgG antibody was reacted for 1 h at 22°C. After a second incubation with HRP-conjugated anti-rabbit IgG goat IgG for 30 min at 22°C, the bound HRP was detected using the ECL Plus Western blot detection system (Pharmacia, Uppsala, Sweden).
Binding Competition Assay with Radio-Labeled C5a Peptide
Peptide fragments of C5a (YSFKDMQLGR) and C5a/RPS19 (YSFKDMQLDRIAGVAAANKKH), which possessed sequences of their second binding sites to C5aR were synthesized as described earlier.8 Tyrosine iodination of the C5a peptide with 125I was carried out using the Iodobead methods (Pierce, Rockford, IL, USA) according to the manufacturer's instruction manual. For binding assay, 125I-C5a peptide was mixed with neutrophils (106 cells/ml) in HBSS containing 20 mM HEPES and 0.5% BSA (pH 7.4) at a putative final concentration 10−7 M in the absence and presence of various concentrations of unlabeled C5a and C5a/RPS19 peptides, respectively.6 The cell suspension mixture was incubated on ice for 60 min and unbound peptides were separated from the cells by centrifugation at 2000 rpm for 2 min at 4°C. After washing the cells thrice in the same medium, radioactivity of bound 125I-C5a peptide on the cells was measured for 2 min in a gamma counter (PerkinElmer, Yokohama, Japan).
Statistical Analysis
The results of a representative examination in at least three different examinations are shown. The experiments were carried out with triplicate samples. Values are expressed as mean±s.d. Statistical significance was assessed using Student's t-test. A P-value <0.05 was considered statistically significant.
RESULTS
C5aR De Novo Synthesis during Apoptosis
To examine the participation of the RP S19 dimer-C5aR system in apoptotic promotion in human HL-60 cells, as has been seen in mouse NIH3T3 cells,11 viable cells were counted by the trypan blue exclusion method at various time points after the induction of apoptosis by 0.5 mM manganese (II) loading. Simultaneous addition of C5a (10−8 M), C5aRA (10−6 M), the anti-C5aR rabbit IgG (5 μg/ml), or the anti-RP S19 rabbit IgG (5 μg/ml) reduced the apoptotic rate as judged by the number of non-dye permeable cells (Supplementary Figure S1B). This indicates that the RP S19 dimer released from the apoptosis-initiated human cell line enhanced the apoptotic rate through binding to C5aR, as in the case of the mouse cell line.
HL-60 and AsPC-1 cells do not constitutively express the C5aR gene in usual culture conditions. To examine if they synthesize C5aR de novo during apoptosis, total RNA from cells at 0, 1, 4, 6, and 12 h after three different apoptotic inductions was prepared for RT-PCR analysis. Transcription of C5aR started from 4 to 12 h, peaking at 6 h in all conditions (Figure 1a). Cell surface expression of C5aR on HL-60 cells at 12 and 18 h after hyperthermia was observed by FACS and by confocal laser scanning microscope (CLSM) analyses. Two cell groups (group R1 and R2) were separated on the base of the forward side scatter values (inset in Figure 1b). Group R1 cells at both 12 and 18 h seemed to be apoptotic, as they expressed phosphatidylserine measured by annexin V-binding. Of note, most of the phosphatidylserine-expressing cells were C5aR-positive (Mean fluorescent intensity (MFI) by FITC-conjugated anti-C5aR mouse IgG; 5 × 102). C5aR (green dots) and phosphatidylserine (red dots) were observed on the cell surface at 12 and 18 h, although most of these did not co-localize. In contrast, group R2 cells solely expressed C5aR. At 12 h, their MFIs were detected to be 2 × 102 in FACS, but cell surface expression of C5aR was not observed in CLSM (Figure 1b upper right). However, at 18 h, we clearly observed C5aR solely expressed on the cell surface in CLSM (Figure 1b lower right), when MFI was achieved to be 4 × 102 in FACS. These data indicate that the C5aR was expressed before exposing phosphatidylserine on the cell surface after apoptotic inductions. Similar results were also obtained for AsPC-1 cells (data not shown).

C5aR on apoptotic cells. (a) The transcription of C5aR in HL-60 cells at each time point after the apoptotic stimulations was detected by semi-quantitative RT-PCR. (b) The expressions of C5aR and phosphatidylserine on HL-60 cells at 12 (upper) and 18 h (lower) after apoptotic stimulation were analyzed by FACS. Double-positive and C5aR-expressing HL-60 cells in groups R1 and R2 were observed by CLSM ( × 1000). The average of percent of annexin V-positive cells (c) and percent inhibition of total apoptosis (d) were recalculated by individual cell cycle analysis using HL-60 (white columns) and AspC-1 cells (black columns) at 24 h after three different apoptotic stimulations in the presence of C5a (10−8 M), C5aRA (10−6 M), and anti-C5aR rabbit IgG (5 μg/ml). Data are mean values±s.d. (*P<0.05 and **P<0.01).
To examine the role of C5aR, apoptosis was initiated in cells by three different apoptotic stimulations in the presence of C5a (10−8 M), C5aRA (10−6 M), or anti-C5aR rabbit IgG (5 μg/ml). The subset of cell in the G1 phase of the cell cycle and the percentage of phosphatidylserine-expressing cells were measured by FACS analyses, as cells at G1 subset33 as well as the phosphatidylserine-expressing cells are considered to be undergoing apoptosis. The percent inhibition of apoptosis at 24 h after three different apoptotic stimulations was measured (Figure 1c). The percent inhibition in three different apoptotic conditions was then re-calculated and is shown in Figure 1d. The percent of the apoptotic cells as measured by G1 in the cell cycle in the presence of C5a, C5aRA, and anti-C5aR rabbit IgG was 30.4±10.6, 27.4±6.9, and 29.4±8.2 in HL-60 cells, and 55.6±15.4, 30.7±10.3, and 27.4±6.9 in AsPC-1 cells, respectively (data not shown). The percent of cells measured with phosphatidylserine exposure in the presence of C5a, C5aRA, and anti-C5aR rabbit IgG was 25.9±4.7 (P<0.049), 25.4±4.4 (P<0.039), and 26.7±0.7 (P<0.0035) in HL-60 cells, and 26.1±4.4 (P<0.038), 25.8±3.8 (P<0.028), and 26.7±0.6 (P<0.013) in AsPC-1 cells, respectively (Figure 1c). The percent in the control condition was 33.7±1.9 in HL-60 cells and 36.3±3.8 in AsPC-1 cells. Inhibition of apoptosis in percent by the presence of C5a, C5aRA, and the anti-C5aR rabbit IgG was 23.5±10.9, 24.8±9.9, and 20.6±4.2 in HL-60 cells, and 28.1±9.3, 28.9±8.1, and 25.8±7.6 in AsPC-1 cells, respectively (Figure 1d).
These results indicate that both of these cell lines de novo synthesized C5aR under the three different apoptotic conditions, and that a common C5aR-mediated signal, which must be mediated at the receptor by a ligand factor in the apoptotic cell culture supernatants, led to an acceleration of apoptotic rate. As the cross-linked RP S19 dimer commonly emerges in the apoptotic cell culture supernatant in studies in which we have examined this,9, 10, 11 the unidentified ligand of C5aR in the above experiments was assumed to be the RP S19 dimer.
Proapoptosis by the RP S19 Dimer-C5aR System
To confirm the presence of RP S19 in the apoptotic cell culture supernatants and to elucidate the generation process of the dimer, aliquots of the culture supernatants and cell pellets of HL-60 cells and of AsPC-1 cells were sampled at 0, 6, 12, and 24 h after the apoptotic stimulations and analyzed by western blotting with anti-RP S19 rabbit IgG (NY4). An isopeptide bond between Lys122 and Gln137 by the action of transglutaminases in the homodimer formation is maintained in the loading buffer. As shown in Figure 2a, the RP S19 dimer (34 kDa) initially appeared in the cell pellets at 6 h and then emerged in the culture supernatants at 12 h. At this time point, the RP S19 monomer was not detected in the supernatants. Consistent with these immunoblot data, the supernatants at 12 h displayed a strong chemotactic activity for monocytes. The chemotactic activity (263.3±37.9) was diminished by the pretreatment of supernatants with anti-RP S19 rabbit IgG (155.0±21.8, P<0.013) or by the pretreatment of indicator monocytes with C5aRA (141.7±5.8, P<0.0053) (Figure 3d). These results indicate that the cross-linked dimerization of RP S19 occurred intracellularly, and then the dimer was extracellularly released around 12 h after apoptotic stimulations. In accordance with the ligand generation, C5aR (43 kDa) was de novo synthesized as shown in Figure 2a.

RP S19 dimer release from apoptotic cells. (a) RP S19 monomer or dimer and C5aR were detected in the supernatants and cell aliquots of HL-60 and AsPC-1 cells at each time point after apoptotic stimulations by western blot. The average of percent of annexin V-positive cells (b) and percent inhibition of total apoptosis (c) were recalculated by individual cell cycle analysis using HL-60 (white columns) and AspC-1 cells (black columns) at 24 h after three different apoptotic stimulations in the presence of 5 μg/ml control rabbit IgG or anti-RPS19 rabbit IgG. Data are mean values±s.d. (*P<0.05 and **P<0.01).

Anti- or pro-apoptosis by HL-60 transformants. (a) Amino acid sequences of the wild-type and mutant RP S19 derived from the constructs. (b) Transcriptions of HA-labeled RP S19 and native RP S19 were detected in Mock-, RPS19-, and Q137N-HL-60 by semi-quantitative RT-PCR. (c) HA-labeled and native RP S19 productions in each of the HL-60 transformants were analyzed by western blot. (d) The monocyte chemotactic potencies of supernatants of apoptotic Mock-, RPS19-, and Q137N-HL-60 at 24 h after manganese (II) loading were compared with simultaneous presence of control PBS (white column), anti-RP S19 rabbit IgG (black column), and C5aRA (hatched column) in chemotaxis chamber, respectively. (e and f) Apoptotic ratios were calculated by cell cycle analysis using Mock- (white column), RPS19- (black column), and Q137N-HL-60 (hatched column) at each time point after the apoptotic stimulations. Data are mean values±s.d. (*P<0.05, **P<0.01).
As described above, these cells expressed C5aR and released the RP S19 dimer during apoptosis. It was next examined whether the RP S19 dimer-C5aR system was responsible for the apoptotic promotion in the above experimental settings.11 FACS analysis was carried out as described above, except for the presence of 5 μg/ml anti-RP S19 rabbit IgG in place of C5a. As shown in Figure 2b, the percent of the phosphatidylserine-expressing HL-60 cells or AsPC-1 cells at 24 h significantly decreased in the presence of anti-RP S19 rabbit IgG (23.0±3.7, P<0.010 or 24.5±1.2, P<0.0068) when compared with the presence of control rabbit IgG (33.7±1.8 or 36.3±3.8). The percent inhibition of apoptosis was 31.9±8.0 and 32.2±5.0 in HL-60 cells and AspC-1 cells, respectively (Figure 2c) (Similar results are shown in Figure 1d). These results indicate that the RP S19 dimer-C5aR system participates in the apoptosis promotion of these human cells, even though they do not constantly express C5aR as observed earlier in apoptotic mouse cells.11
Cessation of Proapoptosis by Q137N-RP S19-Producing HL-60 Cells
To effectively bind to the C5aR, the intermolecular isopeptide bond between Lys122 and Gln137 is essential when RP S19 molecules are dimerized by the transglutaminase-catalyzed reaction. If an isopeptide was formed between the Lys residue and a Gln residue other than Gln137, the PR S19 dimer could not obtain the proper conformation to bind to the C5aR.10 To confirm the proapoptosis by the RP S19 dimer, we established an HL-60 transformant-producing Gln137Asn mutant RP S19 (Q137N-HL-60). As an inverse experiment, we also established an HL-60 transformant over-producing wild-type RP S19 (RPS19-HL-60) (Figure 3a). The transcripts of the RP S19 gene and the hemagglutinin (HA)-tagged RP S19 artificial genes were analyzed by RT-PCR (Figure 3b). Western blot analysis using an image analyzer with NIH Image 1.63 software showed that the total amount of RP S19 and HA-RP S19 in RPS19-HL-60 was twice the levels as that in Q137N-HL-60 or in Mock-HL-60 (Figure 3c). The production of the recombinant, wild-type, or Q137N HA-RP S19 did not affect the cell growth of the transformants (data not shown). The RP S19 dimer was detected by western blot in each culture medium at 18 h after hyperthermia (data not shown), although Q137N-HL-60 did not exhibit monocyte chemotactic activity (Figure 3d). Thus, we successfully established Q137N-HL-60 that produced a non-functional RP S19 dimer with the cross-linkage between Lys122 and a Gln residue other than Gln137.10
Non-apoptotic viable cells in 96-well tissue culture plates were counted using the trypan blue dye exclusion method at 0, 4, 6, 12, 24, 48, and 72 h after the apoptotic stimulation with hyperthermia. The viability of Q137N-HL-60 (98.18±1.35%) was significantly higher than that of Mock-HL-60 (55.45±3.28%) at 24 h (P<0.0016), and, inversely, that of RPS19-HL-60 (32.73±1.82%) was significantly lower (P<0.00047). Similar results were obtained when the other apoptotic stimulations were used (data not shown). These results were confirmed by the cell cycle analysis using aliquots of Mock-HL-60, RPS19-HL-60, and Q137N-HL-60 cells at 0, 6, 12, and 24 h after the apoptotic stimulations. In the thapsigargin treatment, the G1 subset ratio of Q137N-HL-60 significantly decreased when compared with Mock-HL-60 at 6 h (P<0.0058), 12 h (P<0.0039), and 24 h (P<0.000029) (Figure 3f). Although there were no significant differences at 6 h (P<0.21) and 12 h (P<0.15) in the manganese (II) loading, a significant difference was observed at 24 h (P<0.000064) (Figure 3e). Inversely, the G1 subset ratio of RPS19-HL-60 significantly increased after the manganese (II) loading or the thapsigargin treatment when compared with that of Mock-HL-60 at 6 h (P<0.00014 or P<0.000040), 12 h (P<0.00025 or P<0.000024), and 24 h (P<0.23 or P<0.000018).
We then performed recovery experiments by supplementing the culture medium of Q137N-HL-60 with the RP S19 dimer (10−8 M) or C5a/RPS19 (10−8 M). In the recovery experiments, we stimulated Q137N-HL-60 with the reagents at half-doses of those used in the above experiments to show the apoptotic promotion more significantly (Supplementary Figure S1). As shown in Figure 4a and b left, both in the manganese (II) loading and in the thapsigargin treatment, the G1 subset ratios of Mock-HL-60 were significantly increased by the presence of the RP S19 dimer when compared with those of control PBS at 24 h (P<0.00059 and P<0.00059), 48 h (P<0.00023 and P<0.036), and 72 h (P<0.0098 and P<0.0098). Similar results were obtained when C5a/RPS19 was used as the supplement. In the same apoptotic conditions, the G1 subset ratio of Q137N-HL-60 significantly recovered compared with control PBS at 24 h (P<0.0033 and P<0.00034 with the RP S19 dimer or P<0.0050 and P<0.00022 with C5a/RPS19), 48 h (P<0.0018 and P<0.00022 with the RP S19 dimer or P<0.0023 and P<0.00052 with C5a/RPS19), and 72 h (P<0.00021 and P<0.00043 with the RP S19 dimer or P<0.0019 and P<0.00035 with C5a/RPS19) in the presence of the RP S19 dimer or C5a/RPS19. C5a/RPS19 as well as the RP S19 dimer fully recovered the G1 subset ratio of Q137N-HL-60 up to the level in Mock-HL-60. Taken together, these data confirm that the RP S19 dimer-C5aR system is a common element in proapoptosis even though C5aR is not constantly expressed in the physiological conditions. It should be also emphasized that C5a/RPS19 was capable of reproducing the RP S19 dimer-mediated apoptosis promotion.

Proapoptosis by the RP S19 dimer. Apoptotic ratios were calculated by cell cycle analysis using Mock- and Q137N-HL-60 at each time point after half-dose of manganese (II) loading (a) or thapsigargin treatment (b) with the control PBS (white column), 10−8 M the RP S19 dimer (black column), and 10−8 M C5a/RPS19 (hatched column), respectively. Data are mean values±s.d. (*P<0.05, **P<0.01).
Anti-Apoptotic and Clearance-Resistant Behaviors of Q137N-HL-60 In Vivo
We have earlier showed that when apoptosis-initiated HL-60 cells by the hyperthermia had been injected into the guinea pig skin, these cells were rapidly cleared by infiltrated monocytes/macrophages without any inflammatory signs, and that the phagocytic apoptotic cell clearance depended on the RP S19 dimer.9, 5 From these earlier observations, we expected anti-clearance behavior of the apoptosis-initiated Q137N-HL-60 in vivo. In the current study, we used Kud:ddY mice because we had recently confirmed the effective ligand function of human RP S19 dimer to the mouse C5aR.11 We fluorescently labeled wild-type-HL-60 in green or Q137N-HL-60 in red, treated with 0.5 mM manganese (II) for 3 h at 22°C, and respectively injected intradermally (107 cells/50 μl) and intraperitoneally (107 cells/50 μl). After 24 h, we observed the fate of injected cells, macroscopically in the case of intradermal injection, and quantitatively by means of FACS for recovered fluorescent cells in the case of intraperitoneal injection. The results were reproducible in three separate experiments, and representative results are shown in Figure 5. Whereas the cell-injected mass disappeared in the case of wild-type HL-60 in the case of our earlier studies,9, 5 it still remained as 6-mm3 mass in the case of Q137N-HL-60 (Figure 5a). It was consistent with the results in FACS analysis for fluorescent HL-60 recovered from the peritoneal cavity. Only 20% of the Q137N-HL-60 was cleared, whereas almost 90% of the wild-type HL-60 was cleared within 24 h (P<0.0021) (Figure 5b and c). It was often seen in CLSM observation that a green-labeled wild-type HL-60 was engulfed by a mouse macrophage as shown earlier in the guinea pig skin9 (Figure 5c-II), whereas it was hardly seen for red-labeled Q137N-HL-60.

Fate of wild-type HL-60 and Q137N-HL-60 in vivo. (a–d) Wild-type HL-60 (red column) and Q137N-HL-60 (green column), which had been labeled with Red and Green Fluorescent SYTO* Nucleic Acid Stains, were treated with 0.5 mM manganese (II) for 3 h, and respectively injected into the skin (107 cells/50 μl) and the peritoneal cavity (107 cells/50 μl) of Kud:ddY mice. After 24 h, the size of remaining skin lesion (denoted by arrows) was measured (a) and the cells recovered from the peritoneal cavities were analyzed by FACS, regarding the red- or green fluorescence and the annexin V-binding characteristic (b and c). The cell morphology was observed by CLSM ( × 1000) (b-I and c-I and II). The number of transplanted non-apoptotic cells remained in the peritoneal cavity was calculated by the following formula; the remaining non-apoptotic HL-60 cell number=the total number of cells in the peritoneal cavity × percent of the large cell size group on the basis of the forward side scatter values × percent of the annexin V-negative and fluorescence-positive cells. These numbers were obtained from three mice in each of the green cell injection and the red cell injection, and analyzed statistically. Data are mean values±s.d. (**P<0.01) (d). Separately, Q137N-HL-60 (e) and wild-type-HL-60 (f) were, respectively, injected into the right or left flank skin of CAnN.Cg Foxn1nu/CrlCrlj (nu/nu) mice. On day 21 after the injection, the size of tumor mass at the injection site (denoted by arrows) was measured under the macroscopic observation.
To obtain direct evidence for the anti-apoptotic capacity of Q137N-HL-60 in the peritoneal cavity, we compared the number of remaining non-apoptotic cells with the fluorescence-positive, but annexin V-binding-negative, characteristics between the red Q137N-HL-60 and the green wild-type HL-60. As shown in Figure 5d, the number of non-apoptotic cells was significantly higher in the red Q137N-HL-60. When the apoptosis-initiated green wild-type HL-60 and red Q137N-HL-60 were intraperitoneally injected together, few number of red Q137N-HL-60 was recovered as in the case of green wild-type-HL-60 (data not shown). We interpreted this as that the RP S19 dimer provided by the apoptotic wild-type HL-60-promoted apoptosis of the Q137N-HL-60.
We examined the anti-apoptotic behavior of Q137N-HL-60 in vivo in another experimental setting, in which we transplanted viable plain cells into the mouse skin and compared the tumorigenic capacity between wild-type HL-60 and Q137N-HL-60. In the long-term experiment, we used nude mice of CAnN.Cg Foxn1nu/CrlCrlj (nu/nu) strain. We injected the same cell number, 107 cells, of the wild-type and the Q137N at the right flank and the left one, respectively, and daily observed the mass formation. In our preliminary experiments, wild-type HL-60 was not transplantable at the cell number injected in the nude mouse strain used (Figure 5f). The results were reproducible in three separate experiments. Representative photo pictures on day 21 are shown in Figure 5. Only Q137N-HL-60 formed the tumor mass at the injected site (Figure 5e). Our interpretation of this result is as follows: because the transplantation of human-derived HL-60 cells into the mouse is a xenotransplantation, apoptosis initiation stress must be given to the xenograft even in the absence of acquired immunity in the recipient nude mouse. The tumorigenic capacity of Q137N-HL-60 at the relatively small transplanted cell number would indicate the resistance of this mutant against the apoptotic stress.
Binding Affinities to C5aR of C5a-Mimicking Peptide and C5a/RPS19-Mimicking Peptide
It is important to elucidate how C5aR mediates opposite cellular responses in apoptosis. Regarding the effects of different receptor ligands, three kinds are known such as the agonist, the antagonist, and the inverse agonist. Among them, the agonist promotes the receptor to mediate intracellular signals, and two others prevent the agonist-induced or autonomic invigoration of the receptor.34, 35 In the case of neutrophil chemotaxis, we have simply thought the effect of the RP S19 dimer to be the C5aR antagonist competing with C5a.3, 24 However, the proapoptotic effect of the RP S19 dimer was inhibited not only by an agonist C5a, but also by an antagonist C5aRA as described above. In addition to this, the C5aR ligand present in the supernatant of apoptotic cells is solely the RP S19 dimer as far as we have studied. These strongly suggested a possibility that the RP S19 dimer functioned as a C5aR agonist in the apoptosis promotion.
Three mechanisms are known to regulate the agonist-induced GPCR function. One is the up- or downregulation of cell surface receptor expression. Second is the regulation by the ligand and third is by RGS. Therefore, when the competition between the RP S19 dimer and C5a in the proapoptosis and anti-apoptosis is concerned, one could assume not only the direct competition at the C5aR ligation, but also an indirect one on the RGS gene expression. The former should be the short while mechanism and the latter should be the long while mechanism.
In accordance with this speculation, one possibility to induce the opposite responses would be different binding affinities between C5a and C5a/RPS19 to C5aR. The molecular differences between C5a and C5a/RPS19 are only two: one is that the latter bears the COOH-terminal elongation with 12 amino acid residues and the other is difference of the carboxyl group important for the second step receptor-binding, the α-carboxyl group of COOH-terminal Arg74 in C5a, and the β-carboxyl group of Asp73 at one residue ahead of the Arg74 in C5a/RPS19.24 C5a, the RP S19 dimer, and C5a/RPS19 ligate C5aR by the two-step binding mechanism.7 The first binding would occur between the NH2-terminal acidic portion of the receptor and a basic cluster in the ligand molecules. The first binding does not directly activate the C5aR, but effectively raises the local concentration of these ligands and thereby promotes the second binding. The second binding occurs between the COOH-terminal -Leu72-Gly73-Arg74-COOH in the case of C5a or -Leu72-Asp-73-Arg74- in the case of C5a/RPS19 and transmembranous interhelical regions of C5aR. The second binding triggers the G protein-dependent signaling. The COOH-terminal elongation of C5a/RPS19 would affect the second binding either by steric hindrance or by affinity enhancement. However, even if the affinity difference was present, it would be hidden by the first binding affinity when we used C5a and C5a/RPS19 as the ligands.36, 37 Indeed, we did not observe difference in the C5aR-binding affinity between C5a and the RP S19 dimer.6 Therefore, we prepared synthetic peptides mimicking the second binding motif of C5a and the motif with the COOH-terminal elongation of C5a/RPS19 such as YSFKDMQLGR and YSFKDMQLDRIAGQVAAANKKH, respectively, and used for the binding affinity analysis. In this experiment, we used neutrophils because of constitutively expressing C5aR. As shown in Figure 6a, whereas these peptides almost equally competed to 125I-labeled C5a peptide in the neutrophil-binding assay at low concentrations, the C5a/RPS19 peptide showed stronger competition capacities than the C5a peptide at high concentrations such as 10−6 and 10−5 M. The difference was small, but significant and reproducible. We replotted these competition curved in logarithmic–logarithmic manner, and calculated apparent binding affinities of C5a and C5a/RPS19 to C5aR from slants of their lines separately at the low and high concentrations (Figure 6a bottom). At low concentrations, the apparent binding affinities of C5a peptide (y=−2145Ln(x)+13516, R2=0.925) and C5a/RPS19 peptide (y=−2023.2Ln(x)+13356, R2=0.9889) were not statistically different. In contrast to this, at the high concentrations, the apparent binding affinity of C5a/RPS19 peptide (y=−3655.2Ln(x)+11123, R2=0.998) was 1.6 times higher than that of C5a peptide (y=−2300.2Ln(x)+11666, R2=0.809). In the case of binding affinity of C5a, there was no statistical difference between at low and high concentrations, indicating the C5a/RPS19 peptide gained an additive binding affinity to C5aR, 60% up from the baseline affinity, at the high concentrations.

RGS3 in neutrophils. (a) The C5aR on neutrophils (106 cells/ml) was competitively ligated by 10−7 M 125I-labeled peptide mimicking the second binding motif of C5a (YSFKDMQLGR) in a simultaneous presence of various concentrations of the unlabeled C5a-mimicking peptide (the dotted line with open circles) or of a peptide mimicking the binding motif with the C-terminus of C5a/RPS19 (YSFKDMQLDRIAGVAAANKKH) (the plain line with closed circles). After incubation for 60 min on ice, the radioactivity of 125I-C5a peptide bound to the cells was measured for 2 min in a gamma counter. Data are mean values±s.d. (b) Transcriptions of RGS1, RGS3, RGS14, and RGS18 were detected in human neutrophils at 4 h after stimulation with chemoattractants by semi-quantitative RT-PCR.
In addition to this, neutrophils pretreated with C5a/RPS19 for 4 h lost their biological activities through both C5aR ligated with C5a and FPR ligated with fMLP without receptor downregulation (Supplementary Figure S2A and B). This cross-inhibition was not explained by the simple receptor competition.
RGS3 De Novo Synthesis Upregulated by the RP S19 Dimer-C5aR System or Downregulated by the C5a-C5aR System
We, therefore, sought an RGS in this family, of which the gene expression was different between the RP S19 dimer and C5a utilizing constantly C5aR-expressing neutrophils, although RP S19 dimer leads the same effect on the C5aR of both neutrophils and apoptotic cells. We prepared the total RNA from fresh peripheral neutrophils at 0, 4, 10, and 24 h after stimulating with C5a/RPS19 (10−8 M), C5a (10−8 M), fMLP (10−8 M), a mixture of fMLP (10−8 M) and C5a (10−8 M), or control PBS, respectively, and performed RT-PCR analyses with specific primer pairs for RGS1, 3, 14, 16, and 18. As shown in Figure 6b, the transcription of RGS3 clearly decreased at 4 h after the stimulation with C5a, fMLP or their mixture, and inversely it increased with C5a/RPS19. In the latter, the increase of RGS3 transcription continued for 10 h, and RGS3 protein also increased after the enhanced transcription (data not shown).
We then examined whether the upregulation of RGS3 gene expression was caused by the C5aR ligation with the RP S19 dimer. Here, we utilized the experimental setting to induce apoptosis in Q137N-HL-60 under the C5a/RP S19 supplement as described above because the timing of the C5aR ligation could be controlled easier. As we had expected, the RGS3 protein expression was enhanced by C5a/RPS19 from 48 to 72 h (Figure 7a). It seemed to be at late phase of apoptosis. FACS analysis using anti-RGS3 rabbit IgG at 48 h after the manganese (II) loading in the presence of C5a/RPS19 classified Q137N-HL-60 into three groups such as RGS3high (MFI: 53.19) with small cell size group, RGS3low (MFI: 17.46) with small cell size group, and RGS3low (MFI: 16.96) with normal cell size group (Figure 7b bottom). The PI staining revealed that the RGS3high with small cell size group (R6 in Figure 7b middle) was at late apoptotic stage with fragmented DNA, that the RGS3low with small cell size group (R5 in Figure 7b middle) was at early apoptotic stage with minimal DNA fragmentation, and that the RGS3low with normal cell size group was still in viable (R2 in Figure 7b upper). In agreement with the FACS analysis, CLSM analysis showed that a hyper-accumulation of cytoplasmic RGS3 (dense green spots) was observed in a late apoptotic cell with fragmented nuclei (double arrow) and less cytoplasmic RGS3 accumulation was observed in an early apoptotic cell with a normal nucleus (single arrow). An apparently normal cell had minimal green spots (arrow head) (Figure 7c).

RGS3 generation during apoptosis. (a) RGS3 was detected in Q137N-HL-60 at each time point after half-dose apoptotic stimulations in the presence of 10−8 M C5a or C5a/RPS19 by western blot. The expression (MFI) and localization (green) of RGS3 were observed in Q137N-HL-60 at 48 h after half-dose apoptotic stimulations in the presence of 10−8 M C5a/RPS19 by FACS (b) and CLSM (c). Early phase apoptotic cells visualized with large nucleus with low PI-positive (arrow head) and middle-size nucleus with high PI-positive (arrow). Late phase apoptotic cell visualized with small nucleus with high PI-positive (double arrow) ( × 1000). (d and e) The expression and localization of RGS3 were observed in RGS3-HL-60 for the same condition without C5a/RPS19.
The RP S19 Dimer-C5aR System-Independent Proapoptosis in RGS3 Over-Expressed HL-60 Cells
The experimental results described above strongly suggest that RGS3 would be a major downstream target of the signal mediated by the RP S19 dimer-C5aR system, and that the constitutive overexpression of RGS3 would cause a proapoptotic state independent of the RP S19 dimer-C5aR system. Therefore, we established an HL-60 transformant that overexpressed RGS3 (RGS3-HL-60) utilizing pCAIN bearing RGS3 cDNA. In the RGS3-HL-60, RGS3 was constitutively expressed, and the amount of RGS3 protein was not affected by the presence or absence of C5a/RPS19 (Figure 8a). We compared the level of RGS3 constitutively produced in the RGS3-HL-60 with the hyper-accumulation induced by the RP S19 dimer-C5aR system in Q137N-HL-60 under the apoptotic condition. Before the manganese (II) loading, the MFI of RGS3 in the RGS3-HL-60 was 28.16 (Figure 7d bottom). At 48 h after the stimulation, the MFI of RGS3 in the late phase apoptotic cells increased to 59.03, whereas those in viable stage did not change. By CLSM (Figure 7e), the hyper-accumulation of RGS3 in RGS3-HL-60 was observed similarly to that in Q137N-HL-60 stimulated with C5a/RPS19. Similar results were obtained in the thapsigargin treatment (data not shown). The G1 subset ratios of RGS3-HL-60 significantly increased after manganese (II) loading or thapsigargin treatment compared with Mock-HL-60 at 24 h (P<0.58 or P<0.047), 48 h (P<0.033 or P<0.0012), and 72 h (P<0.036 or P<0.72) (Figure 8b). It is important that the apoptotic rate in the RGS3-HL-60 was not affected by the presence of C5a/RPS19 or of the anti-RP S19 rabbit IgG.

Proapoptosis by RGS3-HL-60. (a) RGS3 was detected in RGS3-HL-60 at each time point after half-dose apoptotic stimulations in the presence or absence of 10−8 M C5a/RPS19 by western blot. (b) Apoptotic ratios were calculated by cell cycle analysis using Mock- (white column) and RGS3-HL-60 (black column) at each time point after half-dose apoptotic stimulations in the presence of 10−8 M C5a/RPS19 (hatched column) or 5 μg/ml anti-RPS19 rabbit IgG (dotted column). Data are mean values±s.d. (*P<0.05, **P<0.01).
Cessation of Proapoptosis by Knock Down of RGS3 in HL-60 Cells
The results with overexpressed RGS indicate that the proapoptotic function of the RP S19 dimer-C5aR system is executed by RGS3 hyper-accumulation. To confirm this, we established an RGS3 knock-down HL-60 transformant that constitutively expressed RGS3 shRNA (RGS3low-HL-60). RGS3low-HL-60 resulted in about a 90% reduction of RGS3 production (Figure 9a). The cell cycle pattern of RGS3low-HL-60 did not change, but their growth rate was about two-thirds the rate of control shRNA-HL-60 (RGS3cont-HL-60). We induced apoptosis by the manganese (II) loading. As shown in Figure 9c, the G1 subset ratios of RGS3low-HL-60 significantly decreased when compared with RGS3cont-HL-60 at 6 h (P<0.0027), 12 h (P<0.046), and at 24 h (P<0.0019). Representative data at 6 h are shown in Figure 9d. The G1 subset ratio in RGS3low-HL-60 was clearly reduced when compared with RGS3cont-HL-60. Similar results were obtained using the thapsigargin treatment (data not shown).

Anti-apoptosis by RGS3 knock down HL-60. (a) RGS3 production was detected in constitutively expressed control shRNA- (RGS3cont-) and RGS3 shRNA- (RGS3low-) HL-60 by Western blot. (b) The constitutive active GPCR-mediated ERK signals were detected in RGS3low Mock-, and constitutively expressed RGS3- (RGS3-) HL-60 by western blot. (c and d) Apoptotic ratios were calculated by cell cycle analysis using RGS3cont- and RGS3low-HL-60 at each time point after manganese (II) loading. Representative cell cycle result at 6 h in RGS3low-HL-60 (thick line) compared with RGS3cont-HL-60 (thin line). Data are mean values±s.d. (*P<0.05, **P<0.01).
Proapoptosis as Consequence of ERK Pathway Inhibition by Accumulated RGS3
Our hypothesis is that the primary proapoptotic mechanism is the hyper-accumulation of RGS3, mediated by the RP S19 dimer-C5aR system, which leads to a reduction of constitutively active GPCR-mediated signals. This proapoptotic mechanism caused by the hyper-accumulation of RGS3 fits well with recent evidence for negative regulations of protein kinase cascades through GPCRs.38 It is likely that the hyper-accumulation of RGS3 can lead to a broad cross-inhibition of G protein-dependent signals through constitutively active GPCRs. We present evidence for this possibility in the following three experiments. Neutrophils pretreated with C5a/RPS19 for 4 h lost the fMLP-induced chemoattraction and respiratory burst reaction in addition to losing the C5a-induced ones (Supplementary Figure S2B and C). We expected that neutrophils incubated for 4 h increased the production of RGS3 by the C5a/RPS19-C5aR system. To confirm this, we prepared neutrophil-like cells differentiated from RGS3-HL-60 by 1.25% dimethyl sulfoxide for 7 days. As might be expected, FPR-mediated cytoplasmic Ca2+ influx and chemoattraction were similarly reduced, as measured by a decrease in Extracellular signal-Regulated Kinase (ERK) phosphorylation, to C5aR-mediated ones (Supplementary Figure S3). In contrast, the C5a-C5aR system-mediated inhibition of apoptosis was further improved by a simultaneous supplementation of fMLP (Supplementary Figure S4).
We examined a possibility that regulation by RGS3 of a protein kinase system would be involved in the cell survival, such as the ERK pathway and the Akt pathway. As shown in Figure 10b, the levels of RGS3 and of phosphorylated ERKs, but not of phosphorylated Akts (data not shown), were inversely correlated in the setting of RGS3 knock down and overexpression. Consistently, the phosphorylated ERKs were weaker in the RGS3-HL-60 than in other cells, as measured by western blot. To further examine the involvement of ERK phosphorylation, G proteins and downstream target molecules were blocked by following inhibitors: PTX blocking activated G proteins, PD98059 inhibiting ERK, or SB203580 inhibiting p38MAPK. In this experiment, Mock HL-60 was induced into apoptosis by the manganese (II) loading or the thapsigargin treatment in the presence of PTX (100 ng/ml), PD98059 (50 μM), or SB203580 (10 μM). As shown in Figure 10a, at 12 h after the thapsigargin treatment, a mild and a strong inhibition of ERK phosphorylation were observed by PTX and by PD98059, respectively. These effects continued up to 24 h. In contrast to them, SB203580 did not affect ERK phosphorylation. Similar results were obtained in the manganese (II) loading (data not shown).

Proapoptosis by downregulation of ERK signals. (a) The constitutively active GPCR-mediated ERK signals in apoptotic parent HL-60 cells at 12 and 24 h after thapsigargin treatment were detected by western blot. Representative cell cycle result (b) or calculated apoptotic ratio (c) at several time points in the presence of control PBS (thin line (b) or white column (c)) compared with in the presence of PTX (thin dot line (b) or black column (c)), ERK inhibitor (PD98059) (thick line (b) or hatched column (c)), and p38MAPK inhibitor (SB203580) (thick dot line (b) or dotted column (c)) by FACS. Data are mean values±s.d. (*P<0.05, **P<0.01).
Consistent with a decrease in ERK phosphorylation, apoptotic promotion was accelerated in the cell cycle analysis. Representative patterns influenced by these inhibitors in the thapsigargin treatment are shown in Figure 10b. As shown in Figure 10c, the G1 subset ratios of Mock HL-60 in the control at 12 and at 24 h were 19.0±0.8 and 36.2±1.1, respectively. When compared with the control, a significant apoptotic promotion was observed in the PD98059 treatment at 12 h (46.5±3.6, P<0.00021) and at 24 h (61.5±2.1, P<0.000049), whereas in the PTX treatment, it was only detected at 24 h (43.8±3.2, P<0.017). On the other hand, the SB203580 treatment did not affect the cell cycle. The overall contribution of the downregulation of ERK pathway to the full measurement of apoptosis seemed to be about 30% (see Figures 1d and 2c). These results indicate that the downregulation of ERK phosphorylation is a main event of proapoptosis induced by the RP S19 dimer-C5aR system-mediated hyper-accumulation of RGS3.
Pro-Apoptotic Characteristics of RGS3-HL-60 Cells In Vivo
We fluorescently labeled RGS3-HL-60 and Mock-HL-60 with green fluorescence or RGS3low-HL-60 with red fluorescence, treated with 0.5 mM manganese (II) for 3 h at 22°C, and intraperitoneally injected (107 cells/50 μl) into Kud:ddY mice in the same way as Figure 5. After 24 h, we recovered the injected cells from the peritoneal cavities, and quantitatively analyzed by means of FACS. The results were reproducible in three separate experiments, and representative results are shown in Figure 11. Almost 10-fold less numbers of the RGS3-HL-60 remained when compared with the RGS3low-HL-60 or with Mock-HL-60. The clearance rate of the RGS3-HL-60 in 24 h was 99%, whereas the others were 90%. This indicated the pro-apoptotic behavior of the RGS3 overexpressed cells in vivo, as observed in vitro. The RGS3low-HL-60 injected intraperitoneally was not so resistant to the phagocytic clearance as the Q137N-HL-60. It is probably for the reason that apoptotic RGS3low-HL-60 were capable of liberating the functional RP S19 dimer to recruit monocytes/macrophages even though a lower amount, whereas Q137N-RP S19 dimer liberated from the Q137N-HL-60 was not functional.

Fate of RGS3-HL-60 and RGS3low-HL-60 in vivo. (a and b) Mock-HL-60 (white column) and RGS3-HL-60 (black column) or RGS3low-HL-60 (hatched column) (107 cells), which had been labeled with Red and Green Fluorescent SYTO* Nucleic Acid Stains, respectively, were treated with 0.5 mM manganese (II) for 3 h, and injected into the peritoneal cavity of Kud:ddY mice. After 24 h, cells were recovered from the peritoneal cavities, and analyzed for the red- or green fluorescence activity and for the annexin V-binding characteristic by FACS. The number of non-apoptotic cells remained in the peritoneal cavity was calculated by the following formula as in the case of Figure 5d. These numbers were obtained from three mice in each of three experimental groups. Data are mean values±s.d. (**P<0.01).
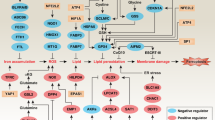
From all the results presented, we conclude that an apoptotic microenvironment leads even cells that sporadically express C5aR to express C5aR and to generate its ligand, the RP S19 homodimer. The RP S19 dimer-dependent agonistic signal through C5aR on apoptosis-initiated cells induces a hyper-accumulation of RGS3 and reduces the constitutively active GPCR-mediated ERK signal. Apoptosis-initiated cells select a shorter life span through the reduction of ERK signal (Figure 12c). Conversely, apoptosis-initiated cells select a longer life span through the enhancement of ERK signal induced by the C5a-C5aR system (Figure 12d).

Post-modification in programmed cell death. (a) Homeostasis: Cells are maintained by the constitutively active GPCR-mediated survival signals. (b) Programmed cell death: Cells go into apoptosis through the death pathway. (c) Proapoptosis in the effector phase of programmed cell death: Apoptotic cell-generated RP S19 dimer-C5aR system-mediated accumulation of RGS3 downregulates the constitutively active GPCR-mediated ERK signals. (d) Anti-apoptosis in the effector phase of programmed cell death: C5a competitively inhibited apoptotic cell-generated RP S19 dimer-C5aR system, and phosphorylated RGS3 is trapped by 14-3-3 molecules,39 resulting in the upregulation of the constitutively active GPCR-dependent ERK signals.
DISCUSSION
Our experimental results suggest that the de novo synthesis of C5aR could be a common event in apoptosis-initiated cells, even though they do not constantly express the C5aR gene (Figure 1). Literature reports relevant to the C5aR gene provide support for our results and interpretation. The promoter region of the mouse C5aR gene possesses a CCAAT sequence, which can be activated by the binding of nuclear factor Y (NF-Y).40 We have currently determined that the CCAAT box is present in the promoter region of the human C5aR gene as well. NF-Y recruits RNA polymerase II and induces transcription of CCAAT box-containing genes.41 A close correlation between the NF-Y-CCAAT box system and apoptosis has been independently reported by several research groups. In the histone deacetylase inhibitor-induced apoptosis or in the genotoxic stress-induced apoptosis, the vitamin D3-upregulated protein 1/thioredoxin-binding protein-2 gene and the Gadd45 gene, both of which contain the CCAAT sequence in their promoter regions, were shown to be expressed.42, 43 In addition, the histone deacetylase inhibitor-induced transforming growth factor β type II receptor gene expression through the activation of NF-Y was described in pancreatic cancer cell lines.44 These genomic analyses suggest that NF-Y could be synthesized or activated during apoptosis and subsequently promote C5aR gene expression in the variably C5aR gene-expressing cells such as HL-60 and AsPC-1 cells. However, this is still a hypothesis that should be experimentally confirmed.
In the case of cells sporadically expressing C5aR, such as HL-60 and AsPC-1 under apoptotic conditions, their life spans could change to be either shorter on supplementation with the RP S19 dimer or longer on supplementation with C5a (Figures 1d and 2c). This is of utmost importance, as it is usually thought that the process of apoptosis or programmed cell death was invariant (Figure 12b). Now, we must consider that, when cells are faced with an apoptotic stimulus, namely, apoptosis-initiated, they would select a shorter or longer life span on the basis of ligands of GPCRs in their microenvironment (Figure 12c and d). This may be valuable from a medical point of view, as it suggests the potential to artificially regulate the apoptosis-initiated cell life span by using the appropriate ligands for apoptosis-promoting or -inhibiting C5aR, and possibly for some other GPCRs. In our case, the ligands are C5a and the RP S19 dimer.
In the case of neutrophils and T cells constitutively expressing C5aR, it has been shown that the C5a-C5aR system leads to the survival of these leukocytes.45 Our current work broadens the phenomenon in leukocytes to various cell types undergoing apoptosis. There is a report that the delay of neutrophil apoptosis is mediated through the ERK pathway, in part,46 making an agreement to our present report. Regarding the regulation of the ERK pathway, we observed the suppression and enhancement of RGS3 transcription at 4 h after stimulation with C5a/RPS19 and C5a, respectively (Figure 6b). Given the increase in RGS3 gene expression, the effect of C5a/RPS19 should be categorized as C5aR agonism. We speculate that the RP S19 dimer released from apoptotic neutrophils at acute inflammatory sites would participate in shortening their life span to limit their effect and resulting inflammation. In contrast to this, C5a may expand their life span to work maximally. In accordance with our opinion, many researchers focus on anti-apoptotic effects of chemoattractants on neutrophils.13, 46 We plan to examine the contribution of the RP S19 dimer to homeostasis of neutrophils at acute inflammatory sites using C5a/RPS19.17
It is mysterious how C5a and the RP S19 dimer or C5a/RPS19 guide the opposite cellular responses in apoptosis even though these molecules ligate C5aR as the agonist. We do not know the mechanism to cause the difference at this moment. The difference of C5a/RPS19 from C5a is the 12-residue elongation at the COOH-terminal with the Gly73Asp substitution. We observed the higher binding affinity of the C5a/RPS19 mimicking peptide bearing the 12-residue elongation than the C5a mimicking peptide to C5aR. The difference of binding affinity was small and visible only at high concentrations (Figure 6a). We do not know at this moment what the apparent affinity difference means. One possibility would be that the 12-residue elongation of the C5a/RPS19 mimicking peptide would bind to another molecular region of the C5aR with a relatively low affinity. Another possibility would be the presence of a molecule beside C5aR, with which the 12-residue elongation interacts at the high concentrations. In either cases, the 12-residue elongation seems to modify the intracellular signal transduction pathway mediated by the C5aR on neutrophils and the apoptosis-initiated cells. The ligation of C5aR by C5a/RPS19 did not cause the receptor internalization (Supplementary Figure S2A), although the ligation by C5a causes the C5aR internalization through the activation of protein kinase C (PKC).47 This suggests that the C5aR-mediated signal under the guidance of the RP S19 dimer or C5a/RPS19 would not link to the PKC pathway. C5aR is a GPCR, and some GPCRs are known to generate Gβγ-dependent signals in addition to the Gα-dependent signals on the separation of the Gαβγ heterotrimetric complex. It must be worthy to compare with the Gα-dependent and Gβγ-dependent signals on the ligation of C5aR between by C5a and by C5a/RPS19.
The second C5aR named C5L2 is present on neutrophils, immature dendritic cells, adipocytes, fibroblasts, and so on.48, 49, 50 It is interesting enough to note that C5L2 is uncoupled with G proteins, and speculated to modulate functions of C5a and C5a desArg as a decoy receptor in inflammatory sites.48, 50 On the other hand, C5L2 on adipocytes was reported to regulate metabolism of this cell type as the receptor of acylation-stimulating protein, in which a G protein-independent intracellular signal transduction pathway would work.49 Judging from the high affinity of C5L2 to C5a and C5a desArg, the RP S19 dimer and C5a/RPS19 would also interact with C5L2. However, it remains to be experimentally elucidated.
From our results, we speculate that other apparently antagonistic ligands of chemotactic receptors released from apoptosis-initiated cells would have sufficient potency to promote apoptosis. When we searched for ligands of GPCRs in released proteins from aged neutrophils,51 apart from ANXA1, β defensins and cathelicidins that interact with the CCR6 chemokine receptor52 and the low-affinity FPRL-1,53 respectively, were found as apparent chemotactic receptor antagonists. Other candidate antagonists are chymase and azurocidin.54 Their receptors, however, have not been identified yet. We are now very interested in whether the above antagonists released from apoptotic neutrophils promote their own apoptosis as agonists.
This study reveals that the RP S19 dimer generates an agonistic signal through C5aR and promotes apoptosis. We propose that the most important molecular mechanism underlying the RP S19 dimer-induced proapoptosis is RGS3 gene expression. There is currently a theory regarding constitutively active GPCRs. According to this theory, the GPCRs, including C5aR, spontaneously change their conformations between inactive and active forms, even in the absence of their ligands.27 Hence, one could assume that a minor proportion of each GPCR would be always in the active state, and these constitutively active receptors would always mediate survival signals, even though the intensity might be much less than the ligand-dependent signal28 (Figure 12a). This leads us to speculate that the RP S19 dimer-C5aR system-mediated hyper-accumulation of RGS3 would overcome the constitutively active GPCR-mediated ERK signals (Figure 9b). It is likely that a cross-inhibition of other GPCR-mediated ERK signals by the hyper-accumulation of RGS3 is the mechanism in the apparent acceleration of apoptosis induced by the RP S19 dimer through C5aR (Figure 12c and Supplementary Figure S2B–S3).55
We do not have an answer to the question of whether or not ligand-dependent agonistic signals through GPCRs completely rescue cells from apoptosis. Anti-apoptosis exerted by C5a was only 30% (Figure 1d). Nevertheless, we found it to be an interesting possibility that there is an fMLP-dependent agonistic signal through FPR (Figure 6b). The de novo synthesis of RGS3 in neutrophils was similarly suppressed at 4 h after the stimulation with fMLP, as in the case of C5a. Moreover, it is noted that the CCAAT box is also present in the promoter region of the human FPRL1 and two genes (NIM_001462 and NIM_002030). The anti-apoptotic effect of C5a was further improved by a simultaneous supplementation of fMLP (Supplementary Figure S4). Further studies are needed to answer this question.
We believe that the RP S19 dimer-dependent C5aR-mediated apoptotic promotion is common. In the present in vitro experimental settings, the contribution of RGS3 to the apoptotic cell life span was 30% at most (Figures 1d and 2c). Although the contribution of RP S19 dimer with apoptosis in the body is not well understood except for the present in vivo data (Figures 5 and 11), it likely varies depending on the apoptotic microenvironment. From all of the results presented here, with support from the published literature, we show that different ligand-guided C5aR-mediated dual signals are, at least in part, physiologically important to regulate the apoptosis-initiated cell life span.
References
Nishiura H, Shibuya Y, Matsubara S, et al Monocyte chemotactic factor in rheumatoid arthritis synovial tissue. Probably a cross-linked derivative of S19 ribosomal protein. J Biol Chem 1996;271:878–882.
Shi L, Tsurusaki S, Futa N, et al Monocyte chemotactic S19 ribosomal protein dimer in atherosclerotic vascular lesion. Virchow Arch 2005;447:747–755.
Nishiura H, Tanase S, Sibuya Y, et al Determination of the cross-linked residues in homo-dimerization of S19 ribosomal protein concomitant with exhibition of monocyte chemotactic activity. Lab Invest 1999;79:915–923.
Umeda Y, Shibuya Y, Semba U, et al Guinea pig S19 ribosomal protein as precursor of C5a receptor-directed monocyte-selective leukocyte chemotactic factor. Inflamm Res 2004;53:623–630.
Shrestha A, Horino K, Nishiura H, et al Acquired immune response as a consequence of the macrophage-dependent apoptotic cell clearance and role of the monocyte chemotactic S19 ribosomal protein dimer in this connection. Lab Invest 1999;79:1629–1642.
Nishiura H, Shibuya Y, Yamamoto T . S19 ribosomal protein cross-linked dimer causes monocyte-predominant infiltration by means of molecular mimicry to complement C5a. Lab Invest 1998;78:1615–1623.
Shibuya Y, Shiokawa M, Nishiura H, et al Identification of receptor-binding sites of monocyte chemotactic S19 ribosomal protein dimer. Am J Pathol 2001;159:2293–2301.
Shrestha A, Shiokawa M, Nishimura T, et al Switch moiety in agonist/antagonist dual effect of S19 ribosomal protein dimer on leukocyte chemotactic C5a receptor. Am J Pathol 2003;162:1381–1388.
Horino K, Nishiura H, Osako T, et al A monocyte chemotactic factor, S19 ribosomal protein dimer, in phagocytic clearance of apoptotic cells. Lab Invest 1998;78:603–617.
Nishimura T, Horino K, Nishiura H, et al Apoptotic cells of an epithelial cell line, AsPC-1, release monocyte chemotactic S19 ribosomal protein dimer. J Biochem (Tokyo) 2001;129:445–454.
Nishiura H, Tanase S, Shibuya Y, et al S19 ribosomal protein dimer augments metal-induced apoptosis in a mouse fibroblastic cell line by ligation of the C5a receptor. J Cell Biochem 2005;94:540–553.
Yamamoto T . Molecular mechanism of monocyte predominant infiltration in chronic inflammation: mediation by a novel monocyte chemotactic factor, S19 ribosomal protein dimer. Pathol Int 2000;50:863–871.
Perianayagam MC, Barakrishnan VS, King AJ, et al C5a delays apoptosis of human neutrophils by a phosphatidyl inositol 3-kinase-signaling pathway. Kidney Int 2002;61:456–463.
Eastman A . The mechanism of action of cisplatin: from adducts to apoptosis. In: B Lippert (ed). Cisplatin. Chemistry and Biochemistry of a Leading Anticancer Drug 1999. Wiley-VCH: Basel, Switzerland, pp 111–134.
Gonzalez VM, Fuertes MA, Alonso C, et al Is cisplatin-Induced cell death always produced by apoptosis? Mol Pharmacol 2001;59:657–663.
Solito E, Kamal A, Russo-Marie F, et al A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J 2003;17:1544–1546.
Maderna P, Yona S, Perretti M, et al Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2-26). J Immunol 2005;174:3727–3733.
Arur S, Uche UE, Rezaul K, et al Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell 2003;4:592–598.
Rescher U, Danielczyk A, Markoff A, et al Functional activation of the formyl peptide receptor by a new endogenous ligand in human lung A549 cells. J Immunol 2002;169:1500–1504.
Perretti M, Croxtall JD, Wheller SK, et al Mobilizing lipocortin 1 in adherent human leukocytes downregulates their transmigration. Nat Med 1996;2:1259–1262.
Lim LH, Solito E, Russo-Marie F, et al Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: effect of lipocortin 1. Proc Natl Acad Sci USA 1998;95:14535–14539.
Lim LH, Pervaiz S . Annexin 1: the new face of an old molecule. FASEB J 2007;21:968–975.
Revollo I, Nishiura H, Shibuya Y, et al Agonist and antagonist dual effect of the cross-linked S19 ribosomal protein dimer in the C5a receptor-mediated respiratory burst reaction of phagocytic leukocytes. Inflamm Res 2005;54:82–90.
Oda Y, Tokita K, Ota Y, et al Agonistic and antagonistic effects of C5a-chimera bearing S19 ribosomal protein tail portion on the C5a receptor of monocytes and neutrophils, respectively. J Biochem (Tokyo) 2008;144:371–381.
Oubrahim H, Chock PB, Stadtman ER . Manganese (II) induces apoptotic cell death in NIH3T3 cells via a caspase-12-dependent pathway. J Biol Chem 2002;277:20135–20138.
Futami T, Miyagishi M, Taira K . Identification of a network involved in thapsigargin-induced apoptosis using a library of small interfering RNA expression vectors. J Biol Chem 2005;280:826–831.
Milligan G . Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol 2003;64:1271–1276.
Dragovich T, Rudin CM, Thompson CB . Signal transduction pathways that regulate cell survival and cell death. Oncogene 1998;17:3207–3213.
Papazisis D, Zambouli OT, Kimoundri ES, et al Protein tyrosine kinase inhibitor, genistein, enhances apoptosis and cell cycle arrest in K562 cells treated with gamma-irradiation. Cancer Lett 2000;160:107–113.
Matsubara S, Yamamoto T, Tsuruta T, et al Complement C4-derived monocyte-directed chemotaxis-inhibitory factor: A molecular mechanism to cause polymorphonuclear leukocyte-predominant infiltration in rheumatoid arthritis synovial cavities. Am J Pathol 1991;138:1279–1291.
Falk W, Goldwin RH, Leonard EJ . A 48 well micro-chemotaxis assembly for rapid and accurate measurement of leukocyte migration. J Immunol Methods 1980;33:239–247.
Laemmli UK . Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–685.
Konteatis ZD, Siciliano SJ, Van Riper G, et al Development of C5a receptor antagonists: differential loss of functional responses. J Immunol 1994;153:4200–4205.
Greasley PJ, Clapham JC . Inverse agonism or neutral antagonism at G-protein coupled receptors: a medicinal chemistry challenge worth pursuing? Eur J Pharmacol 2006;553:1–9.
Cotecchia S . Constitutive activity and inverse agonism at the alpha1adrenoceptors. Biochem Pharmacol 2007;73:1076–1083.
Pease JE, Burton DR, Barker MD . Generation of chimeric C5a/formyl peptide receptors: towards the identification of the human C5a receptor binding site. Eur J Immunol 1994;24:211–215.
Mery L, Boulay F . The NH2-terminal region of C5aR but not that of FPR is critical for both protein transport and ligand binding. J Biol Chem 1994;269:3457–3463.
Han JI, Huang NN, Kim DU, et al RGS1 and RGS13 mRNA silencing in a human B lymphoma line enhances responsiveness to chemoattractants and impairs desensitization. J Leuko Biol 2006;79:1357–1368.
Niu J, Scheschonka A, Druey KM, et al RGS3 interacts with 14-3-3 via the N-terminal region distinct from the RGS (regulator of G-protein signalling) domain. Biochem J 2002;365:677–684.
Hunt JR, Martin CB, Martin BK . Transcriptional regulation of the murine C5a receptor gene: NF-Y is required for basal and LPS induced expression in macrophages and endothelial cells. Mol Immunol 2005;42:1405–1415.
Kabe Y, Yamada J, Uga H, et al NF-Y is essential for the recruitment of RNA polymerase II and inducible transcription of several CCAAT box-containing genes. Mol Cell Biol 2005;25:512–522.
Hirose T, Sowa Y, Takahashi S, et al p53-independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 2003;22:7762–7773.
Butler LM, Zhou X, Xu WS, et al The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci USA 2002;99:11700–11705.
Huang W, Zhao S, Ammanamanchi S, et al Trichostatin A induces transforming growth factor beta type II receptor promoter activity and acetylation of Sp1 by recruitment of PCAF/p300 to a Sp1.NF-Y complex. J Biol Chem 2005;280:10047–10054.
Lalli PN, Strainic MG, Yang M, et al Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 2008;112:1759–1766.
Perianayagam MC, Barakrishnan VS, Pereira BJG, et al C5a delays apoptosis of human neutrophils via an extracellular signal-regulated kinase and Bad-mediated signaling pathway. Eur J Clin Invest 2004;34:50–56.
Suvorova SE, Gripentrog MJ, Oppermann M, et al Role of the carboxyl terminal di-leucine in phosphorylation and internalization of C5a receptor. BBA 2008;1783:1261–1270.
Gerard NP, Lu B, Liu P, et al An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. J Biol Chem 2005;280:39677–39680.
MacLaren R, Cui W, Cianflone K . Adipokines and the immune system: an adipocentric view. Adv Exp Med Biol 2008;632:1–21.
Monk PN, Scola A-M, Madala P, et al Function, structure and therapeutic potential of complement C5a receptors. Br J Pharm 2007;152:429–448.
Scannell M, Flanagan MB, deStefani A, et al Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol 2007;178:4595–4605.
van den Berg RH, Faber-Krol MC, van Wetering S, et al Inhibition of activation of classical pathway of complement by human neutrophil defensins. Blood 1998;92:3898–3903.
Zanetti M, Gennaro R, Romeo D . Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett 1995;374:1–5.
Chertov O, Ueda H, Xu LL, et al Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J Exp Med 1997;186:739–747.
Anger T, Klintworth N, Stumpf C, et al RGS protein specificity towards Gq- and Gi/o-mediated ERK 1/2 and Akt activation, in vitro. J Biochem Mol Biol 2007;40:899–910.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website (http://www.laboratoryinvestigation.org)
Rights and permissions
About this article
Cite this article
Nishiura, H., Nonaka, H., Revollo, I. et al. Pro- and anti-apoptotic dual functions of the C5a receptor: involvement of regulator of G protein signaling 3 and extracellular signal-regulated kinase. Lab Invest 89, 676–694 (2009). https://doi.org/10.1038/labinvest.2009.27
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2009.27
Keywords
This article is cited by
-
Elevated microRNA-25 inhibits cell apoptosis in lung cancer by targeting RGS3
In Vitro Cellular & Developmental Biology - Animal (2016)
-
The role of the ribosomal protein S19 C-terminus in Gi protein-dependent alternative activation of p38 MAP kinase via the C5a receptor in HMC-1 cells
Apoptosis (2010)
-
Rgs3
AfCS-Nature Molecule Pages (2009)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}