
Abstract
Reactive oxygen species (ROS) formation is associated with inflammation and vasculature dysfunction. We investigated the potential role of the NADPH oxidase on vascular Toll-like receptor (TLR) expression and carotid neointimal formation in high-fat (HF) diet-induced obesity (DIO) model. Using mice DIO and common carotid artery flow cessation-induced lesion formation models, we examined vascular TLR2 and TLR4 expression and neointimal formation in NADPH oxidase subunit p47phox-deficient (p47phox–/–) mice. Feeding C57BL/6J mice an HF diet for 22 weeks resulted in significant increases in p47phox, TLR2 and TLR4 expression in vascular tissues compared with mice fed a low-fat (LF) diet. Minimal changes in TLR2 and TLR4 expression was detected in p47phox–/– DIO mice. Furthermore, flow cessation-induced angiogenic and inflammatory response and neointimal formation were significantly attenuated in p47phox−/− DIO mice compared with wild-type DIO mice. In addition, exposure of endothelial cells to leptin led to ROS formation; this was accompanied by upregulation of TLR2, TLR4 expression and its downstream signaling. Leptin also increased endothelial cell migration and proliferation. Pharmacological inhibition of NADPH oxidase or genetic deletion of p47phox significantly diminished these alterations. Obesity increases neointimal formation via a mechanism involving p47phox–TLRs signaling, suggesting that the NADPH oxidase may represent a potential novel therapeutic target for the treatment of obesity-associated vascular inflammation and dysfunction.
Similar content being viewed by others

Main
Obesity is associated with significantly increased mortality and morbidity from type 2 diabetes and cardiovascular diseases. Although obesity is a well-known independent risk factor for cardiovascular diseases, the mechanisms that relate obesity to vascular endothelial dysfunction is poorly understood. Recently, studies have implicated elevated systemic oxidant stress as an important mechanism linking obesity and the increased risk of atherosclerosis.1, 2 Deletion of the p66shc longevity gene to reduce systemic and tissue oxidative stress has been shown to attenuate early atherogenesis in mice fed a high-fat (HF) diet.3 Furthermore, treatment with an NADPH oxidase inhibitor to reduce reactive oxygen species (ROS) formation has been shown to attenuate the abnormal production of adipocytokines and tumor necrosis factor-α (TNF-α) in adipose tissue.4
The multiprotein complex NADPH oxidase, originally identified in phagocytes, is now known to be expressed in endothelial cells. The NADPH oxidases are major sources of ROS in vascular cells and many homologies of the leukocyte NADPH oxidase complex, including p22phox, p47phox, p67phox and gp91phox have been identified in endothelial and vascular smooth muscle cells (VSMCs). Toll-like receptors (TLRs), a family of pattern recognition receptors, is important in the innate immune system. Activation of TLR is linked to the expression of proinflammatory cytokines such as TNF-α and the activation of nuclear factor-κB (NF-κB) signaling pathways in several cell types.5, 6 TLRs are expressed in many cell types including macrophages, adipocytes and skeletal muscle cells. Recently, TLR4 expression has been found in adipocytes isolated from ob/ob and diet-induced obese mouse models, and has been implicated to be involved in the obesity-induced activation of inflammatory and metabolic signaling in insulin resistance.7, 8, 9 TLRs are expressed in the endothelial cells and are upregulated by NADPH oxidase.10, 11 So far, the role of NADPH oxidase on vascular TLR signaling and neointimal formation in obesity has not yet been explored.
Leptin, a protein encoded by the obese (Ob) gene, exerts inflammatory and angiogenic effects on endothelial cells and contributes to vascular dysfunction.12, 13, 14, 15 Obesity is characterized by variable degrees of hyperleptinemia, inflammation and elevated levels of adipocytokines. In addition, leptin has been shown to contribute to the intima–media thickness in obesity.12, 13, 14, 15 A recent study has revealed that treatment with leptin enhances neointimal lesion formation.16 The role of NADPH oxidase in leptin-induced ROS formation, inflammation and endothelial cell dysfunction are not fully understood.
Using p47phox-deficient mice and the HF diet-induced obesity (DIO), we have investigated the essential role of NADPH oxidase on vascular TLR expression and neointimal lesion formation. In addition, we have examined whether leptin-induced vascular ROS formation, inflammation and angiogenesis are dependent on NADPH oxidase.
MATERIALS AND METHODS
Diet-Induced Obesity Model
C57BL/6J mice (n=30) were purchased from Jackson Laboratory. The p47phox knockout mice (n=30) were originally provided by Dr Steve Holland at NIAID. The p47phox knockout mice (C57BL/6J background) were back breeding for 10 generations. The experimental mice (15 mice/each group) were fed with either a normal chow diet or an HF diet (D12451i 45% kcal diet, Research Diets Inc., NJ, USA) for 22 weeks.
Mouse Common Carotid Ligation Model
Beginning at 22 weeks, mice common carotid artery lesions17, 18 were induced by ligation of the left common carotid artery. At 28 days after surgery, the experimental animals were euthanized to evaluate carotid artery neointimal formation.
Quantification of Neointimal Formation
The carotid arteries were sectioned and stained with hematoxylin and eosin (HE). The neointima and media images were captured at × 10 magnification with a digital camera. Neointima and media areas were measured using Image-Pro Plus software (Media Cybernetics).14, 19
Neointimal CD31 and ICAM-1 Immunostaining
Carotid arteries were sectioned (10 μm) and incubated with FITC-monoclonal anti-mouse CD31 antibody (1:50; Sigma, MO, USA) or ICAM-1 antibody (1:50; Santa Cruz, CA, USA). Staining for CD31 was examined using confocal microscopy.20, 21 The neointimal neovascularization was expressed as total CD31-positive cells per mm2. Staining for ICAM-1 was visualized with diaminobenzidine substrate. Sections without primary antibodies were used as control.
Western Immunoblots for p47phox, TLR2, TLR4, TNF-α, Ang-2, MyD88 and IRAK-4
Tissue and cell lysates were subjected to SDS–PAGE and transferred to a nitrocellulose membrane. The membranes were immunoblotted with anti-p47phox or interleukin-1 receptor-associated kinase-4 (IRAK-4) (1:1000; Cell Signaling Technology, MA, USA), polyclonal rabbit TNF-α, Ang-2 or polyclonal goat TLR2, TLR4, MyD88 (1:1000; Santa Cruz).
Experimental Protocols
Before exposure to 50 ng/ml leptin (recombinant human leptin; R&D System Inc., MN, USA), mouse heart microvascular endothelial cells (MHMECs) were pretreated for 30 min with the NADPH oxidase inhibitors, diphenylene iodinium (DPI, 10 μM; CalBiochem, San Diego, CA, USA) and apocynin (Apo, 200 μM; CalBiochem) or the superoxide dismutase (SOD) mimetic, 4-hydroxy-TEMPO (Tempol, 5 mM; Sigma).
Measurement of NADPH Oxidase Activity and ROS
NADPH oxidase activity was determined by lucigenin-dependent chemiluminescence using a luminometer.22, 23 ROS formation was determined using a probe chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA; Molecular Probes Inc., OR, USA), as previously described.22, 23
p47phox Membrane Translocation and Binding to gp91phox
For p47phox membrane translocation studies, cell lysates were centrifuged at 10 000 r.p.m. for 10 min. The supernatant was recentrifuged at 100 000 r.p.m. for 24 h. The pellets were centrifuged at 12 000 r.p.m. for 30 min, and the membrane fraction was isolated.24 The membrane fraction was then detected with anti-p47phox antibody by western blot analysis. For p47phox binding to gp91phox assay, cell lysates were immunoprecipitated with p47phox antibody (2 μg/mg of total cell protein) for 16 h at 4°C, followed by 2 h of incubation with 1:1 protein A:protein G-sepharose. The immunoprecipated lysates were separated using SDS–gel electrophoresis. After transfer to a nitrocellulose membrane, the blot was incubated with anti-mouse gp91phox (1:1000; Santa Cruz) for 1 h. The membrane was washed and incubated with secondary antibody coupled to horseradish peroxidase.
Cell Migration Assay
Migration assays were performed as previously described.22, 23 Polycarbonate filter wells were coated with type I collagen and MHMEC were plated in the upper chamber. The cells were then allowed to migrate for 24 h. Ten randomly selected fields were counted.
Cell Proliferation by MTT Assay
Cell proliferation was assayed using a cell proliferation (MTT) kit according to the manufacturer's instructions (Roche Diagnostic Corp., IN, USA).
All procedures were in conformance with the Institute for Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Statistical Analysis
Statistical analysis was performed using ANOVA followed by t-test corrected for multiple comparisons (Student–Newman–Keuls). Significance was set at P<0.05.
RESULTS
p47phox TLR2 and TLR4 Expression in the Vascular Tissue of the DIO Mouse Model
Feeding mice an HF diet for 22 weeks resulted in a dramatic increase in p47phox expression in the vascular tissues of wild-type (WT) DIO mice (Figure 1a). Next, we sought to determine whether an HF diet increases TLR2 and TLR4 expression and whether the absence of p47phox blocks this effect. Feeding mice an HF diet led to significant increases in TLR2 and TLR4 expression in vascular tissues taken from WT DIO mice compared with control mice fed a low-fat (LF) diet (Figure 1b and c). Minimal changes in TLR2 and TLR4 expression were detected in p47phox–/– DIO mice (Figure 1b and c). As shown in Supplementary data 1A and B, feeding WT and p47phox−/− mice an HF diet led to a gradual increase in body weight and fat tissue deposit. Interestingly, the HF-induced body weight gain and fat tissue deposit were less in p47phox−/− DIO mice compared to WT DIO mice.

Expression of p47phox, TLR2 and TLR4 in the vascular tissue of the DIO mice. (a) Effect of fed a low-fat (LF) or high-fat (HF) diet on p47phox protein expression in the vascular tissues isolated from WT mice. Densitometric data from western blot analyses showing that an HF diet significantly increases p47phox expression in WT DIO mice compared with an LF diet. (b) Expression of TLR2 in the vascular tissues of WT or p47phox mice fed an LF or an HF diet. Western blot densitometric data revealing that an HF diet significantly increases TLR2 expression in WT DIO mice (black bars) compared with LF diet mice (white bars), whereas an HF fails to induce TLR2 expression in p47phox−/− mice. (c) Expression of TLR4 in the vascular tissues isolated from WT or p47phox−/− mice fed an LF or an HF diet. Densitometric data demonstrating that an HF diet significantly increases TLR4 expression in WT mice fed an HF diet (black bars) compared with LF diet (white bars). An HF had little effect on TLR4 expression in p47phox−/− mice (mean±s.d., n=7–8 mice, *P<0.05).
Deficiency of p47phox Attenuates Carotid Artery Flow Cessation-Induced Neointimal Lesion Formation in the DIO Mouse Model
To investigate the effect of NADPH oxidase on vascular lesion formation, we used carotid artery ligation as a model for neointimal formation in DIO mice. No neointimal lesion was developed in both p47phox−/− and WT carotid arteries after 28 days ligation in the LF-fed mouse model (Figure 2a). A larger neointimal lesion was present in WT DIO mice carotid arteries 28 days after ligation. The neointima was significantly smaller in p47phox−/− DIO carotid arteries (Figure 2b). In addition, neointimal area in the p47phox−/− DIO mice was significantly reduced compared with WT DIO mice (Figure 2c). However, media area had no significant changes both in WT and p47phox−/− DIO mice (data not shown).


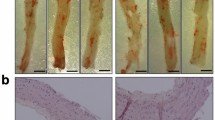
Carotid artery flow cessation-induced neointimal formation in the wild type (WT) and p47phox−/− DIO mice. (a) Representative images of cross sections of carotid artery subjected to flow cessation in WT and p47phox−/− mice fed an LF diet. (b) Representative cross sections of carotid artery flow cessation-induced neointimal lesion formation in WT and p47phox−/− DIO mice. Cross sections were stained with hematoxylin and eosin (HE). (c) Quantitative analysis of neointiamal area in the carotid arteries at 28 days after ligation in DIO mice model. Data are expressed as area of neointimal per mm2 (mean±s.d., n=7–8 mice, *P<0.05, compared with WT DIO mice).
Deficiency of p47phox Attenuates Carotid Artery Flow Cessation-Induced Neointimal Angiogenic and Inflammatory Response in the DIO Mouse Model
To determine whether NADPH oxidase promotes neointimal lesion formation via triggering angiogenic and inflammatory cascades, we examined Ang-2 and TNF-α expression in DIO mice. Ang-2 and TNF-α expression were significantly increased in the carotid of WT DIO mice, but not in DIO mice lacking p47phox (Figure 3a and b). Furthermore, our morphological analyses showed a robust inflammatory and angiogenic response in the neointima of WT DIO mice, confirmed by consistent detection of numerous structures positive for an endothelial-specific marker CD31 (Figure 3c) and ICAM-1 (Figure 3d) in the neointima. Importantly, intralesion inflammatory and angiogenic response was significantly reduced in the p47phox−/− DIO mice (Figure 3c and d).

Carotid artery flow cessation-induced neointimal angiogenic and inflammatory response in the wild type (WT) and p47phox−/− DIO mice. (a) Representative western blot analysis of Ang-2 expression in the vascular tissue of WT DIO mice and p47phox−/− DIO mice. Densitometric data demonstrating that WT mice fed an HF diet (black bars) results in a significant increase in Ang-2 expression compared with LF diet (white bars), whereas an HF diet fails to induce Ang-2 expression in p47phox−/− mice (mean±s.d., n=7–8 mice, *P<0.05). (b) Western blot densitometric data demonstrating that WT mice fed an HF diet (black bars) results in a significant increase in TNF-α expression compared with LF diet (white bars), whereas an HF diet fails to induce TNF-α expression in p47phox−/− mice (mean±s.d., n=5 mice, *P<0.05). (c) (Left) Upper panel: representative cross section of the carotid artery of WT DIO mice at 28 days after surgery staining with the endothelial cell marker, CD31. Upper right inset illustrates a × 40 magnification of one of numerous CD31-positive structures (green, boxed) detected within the neointima (arrows point to some of these). Bottom panel: representative cross section of the carotid artery of p47phox−/− DIO mice staining with CD31. (Right): Quantitative analysis of angiogenic response in the carotid arteries. Data are expressed as total CD31-positive cells per mm2 (mean±s.d., n=5 mice, *P<0.05, compared with WT DIO mice). (d) Deficiency of p47phox attenuates carotid artery ligation-induced inflammatory response in DIO mice. (Left) Upper panel: representative cross section of the carotid artery of WT DIO mice at 28 days after surgery staining with ICAM-1. Bottom panel: representative cross section of the carotid artery of p47phox−/− DIO mice staining with ICAM-1. Right: ICAM-1-positive cells in the neointima were significantly reduced in p47phox−/− DIO mice. Data are expressed as total ICAM-1-positive cells per field (10 × ) (mean±s.d., n=5 mice, *P< 0.05, compared with WT DIO mice).
Expression of Ang-2 and p47phox are Upregulated in Atherosclerotic Neointima of ApoE Deficiency (ApoE KO) Mice
ApoE KO mice fed an HF diet for 24 weeks demonstrated a significant increase in atherosclerotic neointima formation as shown in (Figure 4a and b). Our western blot analysis further demonstrated that p47phox and Ang-2 protein expression in atherosclerotic aorta was upregulated during atherosclerotic lesion progression (Figure 4c).

Neointimal formation, Ang-2 and p47phox expression in high fat-diet fed ApoE KO mice. (a and b) Representative cross sections of aorta atherosclerotic neointimal lesion formation in ApoE deficiency (ApoE KO) mice fed a high-fat diet. Cross sections were stained with hematoxylin and eosin (HE). (c) The expression of p47phox and Ang-2 in C57BL/6J (WT) and ApoE (ApoE KO) mouse aorta. p47phox and Ang-2 expressions were increased in the advanced lesion of ApoE−/− mouse aorta.
Leptin Stimulates ROS Formation via Activation of p47phox
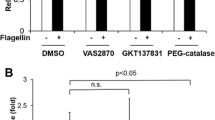
Exposure of WT MHMEC to leptin (50 ng/ml) resulted in p47phox membrane translocation and binding to NADPH oxidase membrane subunit gp91phox (Figure 5a and b). Furthermore, exposure of MHMEC to leptin led to an increase in ROS formation (Figure 5c and d). As shown in Figure 5c and d, pretreatment with Apo (200 μM) or the SOD mimetic, tempol (Temp, 5 mM), for 30 min significantly inhibited leptin-induced ROS formation. Deficiency of p47phox abolished leptin-induced ROS formation (Figure 5c).

Effect of leptin on NADPH oxidase subunit p47phox activation and ROS formation in microvascular endothelial cells. (a) Stimulation of WT MHMEC with leptin for various times up to 60 min caused a gradual increase in p47phox membrane translocation. The figures are representative of an experiment that was repeated at least three times with similar results. (b) Stimulation of WT MHMEC with leptin for 30 min resulted in an increase in p47phox binding to gp91phox. The figures are representative of an experiment that was repeated at least three times with similar results. (c) Effects of the NADPH oxidase inhibitor, SOD mimetic and deficiency of p47phox on leptin-stimulated ROS formation in MHMEC. Pretreatment of MHMEC with either apocynin (Apo, 200 μM) or the SOD mimetic, tempol (Temp, 5 mM) for 30 min led to suppression of leptin-induced ROS formation. Leptin failed to stimulate ROS formation in p47phox−/− MHMEC. (d) Pretreatment of MHMEC with either Apo (200 μM) or Temp (5 mM) for 30 min resulted in significant inhibition of leptin-induced superoxide formation as measured by lucigenin-dependent chemiluminescence. Treatment with Apo or Temp alone had no effect on the basal superoxide production in MHMEC (RLU, relative light units; n=3–4 cell lines, data are mean±s.d., *P<0.05, compared with leptin; #P<0.05, compared with baseline).
Leptin-Induced TLR2 and TLR4 Expression is Dependent on the NADPH Oxidase Subunit p47phox
To determine the associations between leptin-induced ROS formation and TLRs signaling, we examined effects of leptin on TLRs expression and its downstream signaling MyD88 and IRAK-4, using p47phox +/+ and p47phox −/− MHMEC. As shown in Figure 6a, exposure of MHMEC to leptin (50 ng/ml) for various time periods results in a gradual increase in TLR2 and TLR4 protein expression seen at 2–4 h. This was accompanied by dramatic increases in MyD88 and IRAK-4 expression (Figure 6b). In p47phox−/− MHMEC, leptin has little effect on TLRs, MyD88 and IRAK-4 protein expression.

Deficiency of p47phox attenuates leptin-induced TLR2, TLR4 expression and its downstream signaling. (a) Exposure of WT MHMEC to leptin for various times up to 24 h led to an increase in TLR2 and TLR4 expression seen at 2–4 h. Leptin failed to induce TLR2 expression in the p47phox−/− MHMEC. TLR4 protein was undetectable in the p47phox−/− MHMEC. (b) Exposure of WT MHMEC to leptin for various times up to 24 h increased TLRs downstream signaling MyD88 and IRAK-4 expression, whereas leptin failed to induce MyD88 and IRAK-4 expression in the p47phox−/−MHMEC. The figures are representative of an experiment that was repeated at least three times with similar results.
Inhibition of NADPH Oxidase Attenuates Leptin-Stimulated Endothelial Cell Migration and Proliferation
Exposure of MHMEC to leptin (50 ng/ml) for various time periods up to 24 h resulted in a gradual increase in Ang-2 expression. Exposure of p47phox−/− MHMEC to leptin failed to cause a dramatic induction of Ang-2 expression (Figure 7a). Exposure to leptin resulted in a significant increase in MHMEC migration. Treatment with DPI (10 μM), Apo (200 μM) or Temp (5 mM) significantly attenuated leptin-induced cell migration (Figure 7b). Exposure of MHMEC to leptin for 72 h also significantly increased cell proliferation. Treatment with Apo (200 μM) partially inhibited leptin-induced cell proliferation, whereas DPI (10 μM) or Temp (5 mM) completely abolished leptin-induced cell proliferation (Figure 7c).

Pharmacological inhibition or genetic deletion of NADPH oxidase on leptin-stimulated endothelial cell migration and proliferation. (a) Exposure of WT MHMEC to leptin for various times up to 24 h increased Ang-2 expression, whereas leptin failed to induce Ang-2 expression in the p47phox−/− MHMEC. The figures are representative of an experiment that was repeated at least three times with similar results. (b) Treatment of MHMEC with the NADPH oxidase inhibitors, diphenylene iodinium (DPI, 10 μM) and Apo (200 μM), or the SOD mimetic, Temp (5 mM), resulted in significant inhibition of leptin-induced endothelial cell migration as measured by Boyden chamber assay (n=3 cell lines, data are mean±s.d., *P<0.05, when compared with leptin). (c) Treatment of MHMEC with DPI (10 μM) and Apo (200 μM) or Temp (5 mM) resulted in significant inhibition of leptin-induced endothelial cell proliferation as measured by MTT assay. Treatment with DPI, Apo or Temp alone had little effect on cell proliferation (n=4 cell lines, data are mean±s.d., *P<0.05, compared with leptin). (d) Deficiency of p47phox attenuates leptin-induced endothelial cell migration. Leptin-induced cell migration was significantly reduced in p47phox−/− MHMEC (black bars) compared with p47phox+/+ MHMEC (white bars, n=3 cell lines, data are mean±s.d., *P<0.05, compared with p47phox+/+ MHMEC, #P<0.05, compared with p47phox+/+ MHMEC stimulated with leptin). (e) Deficiency of p47phox abolished leptin-induced endothelial cell proliferation. Leptin had little effect on the cell proliferation in p47phox−/− MHMEC (black bars) compared with p47phox+/+ MHMEC (white bars, n=4 cell lines, data are mean±s.d., *P<0.05, compared with p47phox+/+ MHMEC).
Deficiency of the NADPH Oxidase Subunit p47phox on Leptin-Induced Endothelial Cell Migration and Proliferation
Using p47phox−/− MHMEC, we further examined the involvement of p47phox activation on leptin-induced cell migration and proliferation. As shown in Figure 7d, leptin-induced endothelial cell migration was significantly impaired in p47phox−/− MHMEC. Interestingly, deficiency of p47phox completely abolished leptin-induced endothelial cell proliferation (Figure 7e).
DISSCUSSION
In these studies, we have explored the link between obesity and oxidative stress on TLR expression and flow cessation-induced neointimal formation. Our data show that NADPH oxidase and TLR signaling are significantly upregulated in the vascular tissue of DIO mice, and deletion of the NADPH oxidase p47phox abolishes both TLR2 and TLR4 expression in vascular tissue. Deletion of the NADPH oxidase p47phox also blunted flow cessation-induced inflammatory and angiogenic response; this was accompanied by a significant inhibition of neointimal formation in DIO mice. Using p47phox−/− MHMEC, our data demonstrate that leptin upregulates TLRs expression and its downstream signaling via an NADPH oxidase-dependent mechanism. Our studies have provided evidence that NADPH oxidase-derived ROS may be a key component in obesity-induced TLR signaling, vascular inflammation and neointimal formation.
Adipose tissue has been recognized as an active endocrine organ that secretes a variety of proinflammatory cytokines such as leptin, TNF-α and IL-6.25 Recent studies have demonstrated that DIO also leads to activation of the TLR4 and the NF-κB, which may be involved in obesity-induced inflammation and insulin resistance in adipose tissue.7, 8, 9 Free fatty acid-induced expression of TNF-α and IL-6 is attenuated in the adipocytes of TLR4−/− mice.7 Our present studies extend these findings and demonstrate that feeding WT mice an HF diet leads to increases in TLR2 and TLR4 expression in the vascular tissue, suggesting that TLR signaling is also a key mediator of inflammation, thereby linking obesity to vascular inflammation and dysfunction.
Clinical studies have implicated elevated oxidant stress as an important mechanism linking obesity and the increased risk of cardiovascular diseases.1, 2 The NADPH oxidase subunit proteins (gp91phox p22phox and p67phox) are increased in animal models of obesity and in the internal mammary arteries of patients with diabetes4, 26, 27 In obese mice, treatment with apocynin reduces ROS production in adipose tissue, attenuates the dysregulation of adipocytokines.28 Previous studies also implicates that insulin resistance is closely associated with oxidative stress.29, 30 Intake of cream and glucose-stimulated ROS formation is dependent on NADPH oxidase.32, 33 Glucose intake also enhances p47phox expression and increases NF-κB activity.29 Furthermore, treatment with insulin resulted in a significant suppression of p47phox expression in myocardial infarction patients.31, 32, 33 Our present studies further reveals that feeding WT mice an HF diet leads to increases in p47phox expression. Intriguingly, HF diet-induced TLR2 and TLR4 expression are strikingly attenuated in p47phox−/− mice, implicating that obesity-induced TLR signaling is mediated by a mechanism involving NADPH oxidase. Hyperleptinemia has been shown to be closely associated with obesity, insulin resistance and intima–media thickness. Increased inflammation and oxidative stress have been shown to contribute to the detrimental effects of leptin. So far, the intracellular molecular mechanisms underlying the association between leptin and vascular dysfunction are not fully understood. Our data have revealed that exposure of endothelial cells to a high concentration of leptin (>0.5–20 ng/ml normal physiological levels) results in upregulation of TLR signaling, implicating involvement of innate immunity in hyperleptinemia-induced endothelial dysfunction. Our data that deletion of p47phox blunted leptin-induced TLR2 and TLR4 expression and its downstream signaling MyD88 and IRAK-4 expression provide additional intracellular molecular basis between ROS formation and TLR signaling.
Neointimal lesion formation in response to arterial injury is markedly attenuated in NADPH oxidase-deficient mice.34, 35 TLR2 and TLR4 expression are markedly enhanced in human atherosclerotic plaque.36 Using the ApoE−/− mouse model, it has been shown that triggering TLR2 activation dramatically increases atherosclerotic plaque formation.37 Furthermore, atherosclerotic plaque formation is significantly reduced in TLR4−/−/ApoE−/− mice.38 TLR2 and TLR4 signaling is involved not only in the development of atherosclerosis but also in neointimal lesion formation. Arterial injury-induced neointimal lesion formation is significantly less in TLR2−/− or TLR4−/− mice.38, 39, 40 Consistent with these findings, our present studies further provide mechanistic data showing that deficiency of the NADPH oxidase subunit p47phox attenuates neointimal lesion formation and TLR expression in DIO mice, suggesting that obesity associated with neointimal lesion formation is mediated by a mechanism involving activation of NADPH oxidase–TLR signaling pathway.
Obesity is associated with increased intima–media thickness of the common carotid artery and that neointimal lesion formation after vascular injury is enhanced in an HF diet-induced animal model of obesity.14, 19 Angiogenesis has likewise been implicated in neointimal formation.41 Animal models also reveal that angiogenesis is increased after the first 4 weeks of an HF diet.42 Treatment with the angiogenesis inhibitors prevents lesion progression in ApoE−/− mice.21 In contrast, treatment with VEGF stimulates plaque angiogenesis and promotes lesion progression both in ApoE−/− mice and in cholesterol-fed rabbit models.43 There is a notable dearth of knowledge about the signaling processes that link ROS, angiogenesis and neointimal lesion formation. For instance, overexpression of the NADPH oxidase subunit, p22phox has been shown to increase HIF-1α, VEGF expression and intralesional angiogenesis and to promote experimental atheroma progression.44 Our present data demonstrated that deficiency of the NADPH oxidase p47phox attenuates Ang-2 expression and blunts angiogenic response in the neointima of DIO mice. Our data also show that leptin-induced Ang-2 expression, as well as cell migration and proliferation are dependent on NADPH oxidase. These data strongly suggest that NADPH oxidase is a key trigger of neointimal angiogenesis and is essential for the lesion formation in the setting of obesity. In addition, our in vivo studies reveal that neointimal ICAM-1 and TNF-α expression is significantly reduced in p47phox−/− DIO, suggesting a role of inflammation in obesity-induced neointimal formation. Although our present studies demonstrated that carotid ligation induced a significant neointimal formation in DIO mice, we did not observe neointimal formation in control mice as other investigator reported.17, 18 Different mouse strain and genetic background have been shown to have a significant different in the neointimal formation in carotid ligation model.45, 46 Whether these factors contribute to this discrepancy in present studies remains unknown. In addition, fed ApoE KO mice with HF diet resulted in a significant atherosclerotic neointimal formation and this was accompanied by dramatic increases in p47phox and Ang-2 expression.
Although elevated leptin concentrations are associated with the intima–media thickness of the common carotid artery in obese patients and that treatment with leptin promotes neointimal formation,14, 15, 16 our present studies did not detect the correlation between the circulating leptin levels with neointimal thickening in DIO mice model. In addition, the metabolic intracellular mechanisms by which DIO modulates NADPH oxidase activation and ROS formation, whether free fatty acids and insulin resistance in DIO is responsible for these alterations remain unknown. Further studies are warranted to links these missing.
In summary, we have shown that HF DIO activates NADPH oxidase and mediates the expression of TLR in the vascular tissues. Deficiency of the NADPH oxidase subunit p47phox attenuates carotid arterial flow cessation-induced angiogenic, inflammatory response and neointimal formation in DIO mice. Our studies elucidate the importance of NADPH oxidase in obesity-induced TLR signaling and inflammation. Pharmacologic or genetic manipulation of the NADPH oxidase system may represent a novel strategy in the treatment of obesity-associated cardiovascular disease.
References
Morrow JD . Is oxidant stress a connection between obesity and atherosclerosis? Arterioscler Thromb Vasc Biol 2003;23:368–370.
Keaney JF Jr, Larson MG, Vasan RS, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol 2003;23:434–439.
Napoli C, Martin-Padura I, de NF, et al. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci USA 2003;100:2112–2116.
Sonta T, Inoguchi T, Tsubouchi H, et al. Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med 2004;37:115–123.
Andreakos E, Sacre S, Foxwell BM, et al. The toll-like receptor-nuclear factor kappaB pathway in rheumatoid arthritis. Front Biosci 2005;10:2478–2488.
Brown MA, Jones WK . NF-kappaB action in sepsis: the innate immune system and the heart. Front Biosci 2004;9:1201–1217.
Shi H, Kokoeva MV, Inouye K, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006;116:3015–3025.
Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, et al. Loss-of-function mutation in TLR4 prevents diet-induced obesity and insulin resistance. Diabetes 2007;56:1986–1998.
Kim F, Pham M, Luttrell I, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res 2007;100:1589–1596.
Faure E, Thomas L, Xu H, et al. Bacterial lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol 2001;166:2018–2024.
Fan J, Frey RS, Malik AB . TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest 2003;112:1234–1243.
Kougias P, Chai H, Lin PH, et al. Effects of adipocyte-derived cytokines on endothelial functions: implication of vascular disease. J Surg Res 2005;126:121–129.
Ciccone M, Vettor R, Pannacciulli N, et al. Plasma leptin is independently associated with the intima–media thickness of the common carotid artery. Int J Obes Relat Metab Disord 2001;25:805–810.
Schafer K, Halle M, Goeschen C, et al. Leptin promotes vascular remodeling and neointimal growth in mice. Arterioscler Thromb Vasc Biol 2004;24:112–117.
Knudson JD, Dincer UD, Zhang C, et al. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. Am J Physiol Heart Circ Physiol 2005;289:H48–H56.
Bodary PF, Shen Y, Ohman M, et al. Leptin regulates neointima formation after arterial injury through mechanisms independent of blood pressure and the leptin receptor/STAT3 signaling pathways involved in energy balance. Arterioscler Thromb Vasc Biol 2007;27:70–76.
Kumar A, Lindner V . Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol 1997;17:2238–2244.
Kawashima S, Yamashita T, Ozaki M, et al. Endothelial NO synthase overexpression inhibits lesion formation in mouse model of vascular remodeling. Arterioscler Thromb Vasc Biol 2001;21:201–207.
Bodary PF, Gu S, Shen Y, et al. Recombinant leptin promotes atherosclerosis and thrombosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2005;25:e119–e122.
Moulton KS, Vakili K, Zurakowski D, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci USA 2003;100:4736–4741.
Moulton KS, Heller E, Konerding MA, et al. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999;99:1726–1732.
Chen JX, Zeng H, Lawrence ML, et al. Angiopoietin-1-induced angiogenesis is modulated by endothelial NADPH oxidase. Am J Physiol Heart Circ Physiol 2006;291:H1563–H1572.
Chen JX, Zeng H, Tuo QH, et al. NADPH oxidase modulates myocardial Akt, ERK1/2 activation and angiogenesis after hypoxia/reoxygenation. Am J Physiol Heart Circ Physiol 2007;292:H1664–H1674.
Ushio-Fukai M, Tang Y, Fukai T, et al. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res 2002;91:1160–1167.
Hausman GJ, Richardson RL . Adipose tissue angiogenesis. J Anim Sci 2004;82:925–934.
Dobrian AD, Schriver SD, Khraibi AA, et al. Pioglitazone prevents hypertension and reduces oxidative stress in diet-induced obesity. Hypertension 2004;43:48–56.
Guzik TJ, Mussa S, Gastaldi D, et al. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002;105:1656–1662.
Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004;114:1752–1761.
Dandona P, Aljada A, Mohanty P, et al. Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab 2001;86:3257–3265.
Dandona P, Dhindsa S, Ghanim H, et al. Angiotensin II and inflammation: the effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J Hum Hypertens 2007;21:20–27.
Mohanty P, Hamouda W, Garg R, et al. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J Clin Endocrinol Metab 2000;85:2970–2973.
Mohanty P, Ghanim H, Hamouda W, et al. Both lipid and protein intakes stimulate increased generation of reactive oxygen species by polymorphonuclear leukocytes and mononuclear cells. Am J Clin Nutr 2002;75:767–772.
Chaudhuri A, Janicke D, Wilson MF, et al. Anti-inflammatory and profibrinolytic effect of insulin in acute ST-segment-elevation myocardial infarction. Circulation 2004;109:849–854.
Barry-Lane PA, Patterson C, van der MM, et al. p47phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J Clin Invest 2001;108:1513–1522.
Chen Z, Keaney Jr JF, Schulz E, et al. Decreased neointimal formation in Nox2-deficient mice reveals a direct role for NADPH oxidase in the response to arterial injury. Proc Natl Acad Sci USA 2004;101:13014–13019.
Edfeldt K, Swedenborg J, Hansson GK, et al. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation 2002;105:1158–1161.
Liu X, Ukai T, Yumoto H, et al. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis 2008;196:146–154.
Michelsen KS, Wong MH, Shah PK, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA 2004;101:10679–10684.
Shishido T, Nozaki N, Takahashi H, et al. Central role of endogenous Toll-like receptor-2 activation in regulating inflammation, reactive oxygen species production, and subsequent neointimal formation after vascular injury. Biochem Biophys Res Commun 2006;345:1446–1453.
Vink A, Schoneveld AH, van der Meer JJ, et al. In vivo evidence for a role of toll-like receptor 4 in the development of intimal lesions. Circulation 2002;106:1985–1990.
Khurana R, Zhuang Z, Bhardwaj S, et al. Angiogenesis-dependent and independent phases of intimal hyperplasia. Circulation 2004;110:2436–2443.
Herrmann J, Lerman LO, Rodriguez-Porcel M, et al. Coronary vasa vasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercholesterolemia. Cardiovasc Res 2001;51:762–766.
Celletti FL, Hilfiker PR, Ghafouri P, et al. Effect of human recombinant vascular endothelial growth factor165 on progression of atherosclerotic plaque. J Am Coll Cardiol 2001;37:2126–2130.
Khatri JJ, Johnson C, Magid R, et al. Vascular oxidant stress enhances progression and angiogenesis of experimental atheroma. Circulation 2004;109:520–525.
Harmon KJ, Couper LL, Lindner V . Strain-dependent vascular remodeling phenotypes in inbred mice. Am J Pathol 2000;156:1741–1748.
Korshunov VA, Berk BC . Strain-dependent vascular remodeling: the ‘Glagov phenomenon’ is genetically determined. Circulation 2004;110:220–226.
Acknowledgements
This work was supported by grants from the American Heart Association grant 0565196B and NIH grant DK074995 (to JX Chen).
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE
All authors declared no conflict of interests.
Supplementary Information accompanies the paper on the Laboratory Investigation website (http://www.laboratoryinvestigation.org)
Rights and permissions
About this article
Cite this article
Chen, JX., Stinnett, A. Critical role of the NADPH oxidase subunit p47phox on vascular TLR expression and neointimal lesion formation in high-fat diet-induced obesity. Lab Invest 88, 1316–1328 (2008). https://doi.org/10.1038/labinvest.2008.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2008.92
Keywords
This article is cited by
-
Vascular smooth muscle cell-specific Igf1r deficiency exacerbates the development of hypertension-induced cerebral microhemorrhages and gait defects
GeroScience (2024)
-
The Role of Toll-Like Receptors in Diabetes-Induced Inflammation: Implications for Vascular Complications
Current Diabetes Reports (2012)
