
Abstract
Interleukin-4 (IL-4) is overexpressed in liver grafts in a context of severe recurrent hepatitis C, during which the development of fibrosis is dramatically accelerated. In this study, we examined the effects of IL-4 on the activation and collagen production of cultured human intrahepatic (myo)fibroblasts (hIHFs), and investigated the underlying mechanisms. The myofibroblastic nature of cells was evaluated morphologically using activation markers (smooth muscle α-actin, vimentin and prolyl 4-hydroxylase). Quiescent hIHFs were obtained by cell incubation in serum-free medium or cell culture on Matrigel. We first analyzed IL-4 receptor expression, STAT-6 activation by IL-4, and STAT-6 inhibition by an anti-IL-4 antibody or by STAT-6 small-interfering RNA (siRNA) transfection. We then focused on collagen production, using quantitative real-time PCR to analyze the effect of IL-4 on the mRNA expression of collagens I, III and IV, and on collagen levels in supernatants of hIHFs, using the Sircol collagen assay. hIHFs cultured in plastic wells appeared to be morphologically activated. The expression of activation markers was reduced by serum deprivation or culture on Matrigel, and restored by IL-4 incubation. The IL-4 receptor was expressed by hIHFs, and STAT-6 was activated following incubation with IL-4. Both anti-IL-4 antibody and STAT-6 siRNA transfection inhibited this activation. The treatment of hIHFs with IL-4 increased the mRNA expression of collagens I, III and IV (P<0.05) and elevated collagen levels in supernatants (P=0.01 vs untreated cells). Therefore, IL-4 exerts profibrotic effects by activating hIHFs and inducing collagen production and secretion. This effect requires IL4-R binding and STAT-6 activation. IL-4 may thus be involved in accelerated course of fibrogenesis during recurrent hepatitis C.
Similar content being viewed by others


Main
Hepatic fibrosis is an outcome of many chronic liver diseases, such as viral and autoimmune hepatitis, alcohol consumption and biliary obstruction. Prolonged liver injury results in hepatocyte damage, which triggers the activation of hepatic stellate cells (HSCs) and the recruitment of inflammatory cells into the liver.1 Liver fibrosis is characterized by an accumulation of extracellular matrix (ECM), resulting from its increased production and decreased degradation and leading to distorted reconstruction of the liver parenchyma that accompanies liver function impairment during most chronic liver diseases.2 HSC play a crucial role in the development of liver fibrosis by constituting an important source of ECM,3, 4 the major components of which are collagens I, III, IV and V.5 During hepatic fibrogenesis, HSCs are activated or transdifferentiate into myofibroblastic cells that lack cytoplasmic lipid droplets, acquire smooth muscle α-actin (α-SMA) expression, produce ECM and then inhibit its degradation by degrading metalloproteases.6, 7 However, liver myofibroblast populations have been shown to be heterogeneous, and at least two populations of myofibroblasts with fibrogenic potential (HSC and hepatic myofibroblasts) accumulate during chronic liver injury in both rats and humans.8, 9 The stimuli that induce the activation of stellate cells or hepatic fibroblasts, such as cytokines, can derive from injured hepatocytes, endothelial cells, Kupffer cells or infiltrating lymphocytes.10 Transforming growth factor-β (TGF-β) is deemed an important inducer of liver fibrogenesis in rats and humans.11, 12 Cytokines other than TGF-β may also be involved in the fibrotic process.
Liver transplantation (LT) has become an accepted therapy for patients with end-stage liver diseases, including hepatitis C virus (HCV)-related cirrhosis.13, 14 The persistence of HCV and the recurrence of hepatitis C are constant after LT,15 resulting in accelerated progression toward fibrosis and impaired patient and allograft survival.16, 17 The mechanisms underlying accelerated liver fibrosis after LT are poorly understood.18 Interleukin-2 (IL-2) is involved in the progression of liver damage during chronic hepatitis C in immunocompetent patients.19 Following transplantation, immunosuppressive drugs (mainly calcineurin inhibitors) markedly reduce IL-2 production, whereas the production of other cytokines, such as IL-4, is less sensitive to their inhibitory effects.20, 21 We have recently shown that IL-4 is overexpressed in patients with severe recurrent hepatitis C when compared to patients with minimal recurrence and to HCV-negative recipients.22 IL-4 overexpression might therefore be involved in the accelerated fibrosis of hepatic lesions in transplant recipients receiving potent anti-IL-2 immunosuppressive therapy.
IL-4 is a pleiotropic cytokine that controls cell growth, regulates the immune system, exerts anti-inflammatory effects by downregulating T-helper 1 (Th1) cell activity23 and promotes T-cell differentiation toward a Th2 phenotype.24 IL-4 exerts its effects by binding to and stimulating its transmembrane receptor (IL-4R), involving the Janus kinases (JAK)-1 and JAK-3, and the signal transducer and activator of transcription (STAT)-6 pathway.25 In vitro studies have shown that IL-4 can induce fibrosis by activating lung fibroblast proliferation, collagen production and myofibroblast differentiation.26, 27, 28 Similar results have been found with human conjunctival fibroblasts.29 In humans, the progression of idiopathic pulmonary fibrosis is associated with IL-4 production.30 High levels of IL-4 expression have also been found in the fibrotic skin tissues of patients with scleroderma,31 and IL-4 is important in collagen production by human conjunctival fibroblasts.29
IL-4 may impact the course of fibrotic liver damage. Indeed, IL-4 enhances collagen synthesis by nonparenchymal liver cells in vitro.32, 33 IL-4 expression is enhanced in the fibrotic liver of Schistosoma-infected baboons, and anti-IL-4 therapy markedly diminishes hepatic fibrosis in Schistosoma-infected mice.34, 35 We have therefore hypothesized that IL-4 overexpression could contribute to the accelerated progression toward fibrosis that occurs during severe chronic recurrent hepatitis C after LT, by inducing intrahepatic fibroblast activation and collagen overproduction. During this study, we examined the profibrotic effects of IL-4 on human intrahepatic (myo)fibroblasts (hIHFs), and investigated the underlying mechanisms. Our results show that IL-4 induces the activation and increases the collagen production of cultured hIHFs involving the STAT-6 pathway.
MATERIALS AND METHODS
Isolation and Culture of Human Intrahepatic (myo)Fibroblasts
hIHFs were isolated from normal liver tissue obtained from patients undergoing partial hepatectomy for metastases or benign tumors. This procedure complied with the ethical guidelines stipulated by French legislation. Liver specimens collected during major hepatectomy were harvested as distant as possible from the tumor, avoiding the immediate peritumoral zone. The lack of significant histological lesions was verified by histological examination before the liver tissue was used for the study. Dissociation was based on a two-step collagenase perfusion method, the cells then being separated by centrifugation over gradients.36 Visible vessels were perfused first with HEPES–EDTA buffer and then with liver digest (Gibco, Cergy-Pontoise, France) containing 0.05% collagenase, at a flow rate of 10 ml per catheter per min (Masterflex peristaltic pump; Bioblock, France) for 30 min. Liver fragments were gently shaken to free any loose liver cells, and were then filtered and centrifuged. Hepatocytes were isolated from the pellet, and hIHFs from the supernatant. In the suspension, hIHFs were separated from other cells by centrifugation at 1800 r.p.m. for 10 min.37 Cell viability was determined by Trypan blue dye exclusion, and freshly isolated hIHFs were seeded in 5% fetal calf serum (FCS)/DMEM (Gibco) supplemented with penicillin (100 U/ml) and streptomycin (100 μg/ml) and cultured at 37°C in a 5% CO2 atmosphere. The culture medium was replaced 1 day after plating. At confluency, cells were subcultured and maintained in 75 cm2 culture flasks (WVR, Strasbourg, France). Cells were used for experimentation between the second and sixth passages.
Each evaluation was performed using cells obtained from at least six different human livers.
hIHFs Culture Conditions
hIHFs (3 × 105 cells per well in six-well plates) were seeded on plastic and in DMEM (5% FCS) and allowed to attach for 6 h. The cells were then starved of serum for 48 h or cultured for 48 h on Matrigel coated in DMEM (5% FCS) to obtain quiescent cells. Then, in all experiments, the medium was replaced with FCS–DMEM (5% FCS) on both plastic and Matrigel, and the cells were cultured for 20 min, 6, 24, 48 or 72 h without treatment (for controls) or with recombinant human TGF-β (10 ng/ml; R&D Systems, Lille, France), recombinant human IL-4 (5, 10, 50, 100 or 500 ng/ml; R&D systems) or recombinant human IL-4 (50 ng/ml; R&D Systems) plus anti-human IL-4 monoclonal antibody (50 ng/ml; R&D Systems). The cells were then detached from the culture plates by trypsination for future analysis, and the supernatants stored at −20°C until the spectrophotometric analysis of collagen production. For P-STAT-6 analyses, cells were incubated in fresh medium (5% FCS/DMEM) and stimulated with human IL-4 (50 ng/ml) for 20 min; longer periods were allowed for activation or collagen production experiments.
Morphology and Characterization of hIHFs
Immunohistochemical methods
For immunohistochemistry, cells were grown on glass coverslips in six-well plates (2 × 104 cells per well) in 5% FCS/DMEM and then washed three times with PBS and fixed in PBS 4% paraformaldehyde. When grown on normal plates, the cells were resuspended in PBS (Gibco; 2 × 104 cells per ml) and then cytocentrifuged on SuperFrost Plus slides (CML, Nemours, France). These cells were then air-dried overnight at room temperature, fixed for 10 min in acetone and used directly or stored at −20°C until immunohistochemical analysis.
The expression of IL-4R (R&D), phosphorylated STAT-6 (P-STAT-6), α-SMA (Dako, Glostrup, Denmark) or vimentin (Dako) was evaluated using an indirect immunoenzymatic method with alkaline phosphatase/anti-alkaline phosphatase complexes, as previously described.38 For intracellular staining, cells were first permeabilized with 0.1% Triton X-100. Anti-human-IL-4R monoclonal antibody (dilution 1/10), anti-α-SMA (1/50), anti-vimentin (1/50) and anti-P-STAT-6 polyclonal antibody (dilution 1/50; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used as the primary antibody. Mouse anti-rabbit immunoglobulin (IgG, Dako, for polyclonal primary antibody only) and rabbit anti-mouse IgG (Dako) were used as secondary antibodies. The slides were then incubated with alkaline phosphatase/anti-alkaline phosphatase complexes (Dako). Alkaline phosphatase activity was revealed for 20 min in fast-red TR (1 mg/ml) and naphthol phosphate (0.2 mg/ml) solutions (Sigma, Saint Quentin-Fallavier, France) containing levamisole (0.24 g/ml; Sigma). The slides were counterstained with Harris hematoxylin. For negative controls, the primary antibody was omitted or replaced by an irrelevant antibody at the same dilution.
Immunofluorescence analysis
In parallel, CD90 (Dianova, GmbH, Hamburg, Germany), α-SMA (Dako) and vimentin (Dako) were evaluated using an immunofluorescence method. Cells were cultured on glass coverslips in DMEM –(FCS 5%), washed three times with PBS and fixed in PBS with 4% paraformaldehyde for 10 min. After fixation, the cells were washed twice with PBS and permeabilized with PBS containing 0.1% Triton X-100 for 10 min (for intracellular staining only: α-SMA and vimentin). The cells were then incubated with primary fluorescent isothiocyanate (FITC)-conjugated antibodies or not (diluted 1/100 in PBS) for 1 h. After three washes, the cells were incubated with FITC-anti-mouse IgG (diluted 1/500 in PBS; Caltag, Burlingame, CA, USA) for 30 min and then washed three times with PBS. The coverslips were mounted in immumount medium (Shandon, USA) and viewed with a fluorescence microscope (Olympus, FITC filter).
Flow cytometry analysis
The expressions of CD90, CD31, α-SMA, vimentin, prolyl 4-hydroxylase and P-STAT-6 were measured by flow cytometry analysis. hIHFs were released from culture plates by trypsination, centrifuged, resuspended in PBS (3 × 105 cells per ml) and then washed. hIHFs were first permeabilized or not with FACs™ permeabilizing solution (BD Biosciences, Le Pont De Claix, France), then stained with FITC-conjugated mouse anti-human CD90 (1/50; Dianova) for 30 min, or with nonconjugated antibodies: anti-human CD31, anti-human α-SMA, anti-vimentin, anti-prolyl hydroxylase antibody (dilution 1/50, Dako), or mouse anti-human P-STAT-6 antibody (dilution 1/50; BD Pharmingen) for 30 min, followed by incubation with a FITC-labeled goat anti-mouse IgG (dilution 1/100; Caltag), for 30 min. A fluorochrome-conjugated irrelevant isotype (mouse IgG) was used as a negative control. Cell staining was then analyzed using a FACSCalibur flow cytometer (BD Biosciences) and CellQuest software.
Inhibition of STAT-6 by the STAT-6 siRNA Transfection of Cultured Cells
Human IHFs (3 × 105 per well in six-well plates) were incubated overnight in DMEM medium supplemented with 5% FCS to reach 50–70% confluency. Cells were then transfected with 100 nM STAT-6 small-interfering RNA (siRNA, ON-TARGETplus SMARTpool with four siRNA sequences targeting human STAT-6; Dharmacon, Chicago, IL, USA), with GAPDH siRNA (ON-TARGETplus siControl GAPDpool; Dharmacon) or with a negative siRNA control (ON-TARGETplus siControl nontargeting pool; Dharmacon) in the presence of 10 μl Lipofectamine 2000 (Invitrogen SARL, Cergy Pontoise, France) and 1 ml FCS-free growth medium. At 6 h after transfection, 1 ml of DMEM medium with 5% of FCS was added to each well, and 24 h after transfection cells were incubated in serum-free medium for 48 h and further stimulated with human IL-4 (50 ng/ml) for 20 min before the evaluation of STAT-6 activation, or for 48 h to evaluate collagen production and expression.
The efficiency of siRNA transfection was checked by quantitative RT-PCR (as described below) and by western blot assay.
Western Blot Assay
Transfected or nontransfected hIHFs were treated or not with IL-4 (50 ng/ml) for 20 min in six-well plates in 5% FCS/DMEM. The cells were rinsed in cold PBS and lyzed by the addition of Laemmli/1.2% β-mercapto buffer (40 mM Tris-HCl, pH 6.8, 5 mM DTT, 1% SDS, 7.5% glycerol, 0.01% bromophenol blue) to the plates. After scraping and boiling, samples (50 μg of protein) were loaded onto a 10% polyacrylamide gel and transferred into a nitrocellulose membrane. Membranes were saturated in 5% milk PBS containing 0.1% Tween 20 for 1 h and then incubated with the following primary antibodies: rabbit polyclonal anti-STAT-6 antibody (dilution 1:500; Cell Signaling Technology, Danvers, MA, USA); rabbit polyclonal anti-STAT-6 phosphorylated antibody (dilution 1:500; Cell Signaling Technology) and mouse monoclonal anti-GAPDH antibody (dilution 1:2000). Membranes were washed and incubated with peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (dilutions 1:5000; Amersham, Saclay, France). After washing, the membranes were incubated with chemiluminescence reagents (Pierce, Rockford, IL, USA). The resulting autoradiographs were then scanned.
Quiescence of hIHFs
Cells were used between the second and sixth passages, and the experiments were performed on hIHFs rendered quiescent by incubation for 2 days in serum-free medium or seeding on basal membrane-like substrate Matrigel-coated plastic plates (Matrigeltm, Becton Dickinson), using the ‘thin film’ method (50 μl per cm2 of growth surface), according to the manufacturer's instructions.39 Cell quiescence was verified by evaluating the reduction or disappearance of specific activation markers (α-SMA, vimentin and prolyl 4-hydroxylase) using flow cytometry. Vitamin A autofluorescence was evaluated using a fluorescence microscope with filters designed to detect FITC.
Evaluation of hIHFs Apoptosis and Necrosis
Annexin V-FITC was used to detect and quantify early stage apoptosis, and propidium iodide (PI) to detect necrosis (Annexin V-FITC kit; Immunotech, Marseille, France), according to the manufacturer's recommendations. After 48 h of culture, hIHFs were treated with IL-4, TGF-β or C2-Ceramid (20 μM; Sigma) as a positive control for apoptosis.40 Cells were suspended in a binding buffer containing Ca2+ and incubated with 1 μg/ml Annexin V-FTIC and 1 μg/ml PI. Apoptosis and necrosis were measured using the FACSCalibur and CellQuest software. A total of 5000 cells were acquired and analyzed in each sample. FITC-positive and PI-negative cells were considered to be apoptotic, whereas PI-positive cells were deemed necrotic and unstained cells considered as normal viable cells.
Evaluation of hIHFs Proliferation
hIHFs (5 × 104 per well) were seeded in 96-well plates (Nunc A/S; Roskilde, Denmark) and incubated for 48 h in serum-free medium. After 48 h, the cells were treated with IL-4 (5, 10, 50 and 100 ng/ml), TGF-β (10 ng/ml) or PDGF (10 ng/ml; R&D Systems) for 48 h. Cell proliferation was measured after pulsing the cells with [3H]thymidine (1 μCi per well) for the last 16 h of culture. The cells were harvested and radioactivity was counted using a scintillation counter. Absorbance at 550 nm was recorded in each well using an ELISA microplate reader.
Reverse Transcriptase–Polymerase Chain Reaction (RT–PCR) for IL-4R mRNA Detection
Total RNA was prepared using the RNeasy minikit (Qiagen SA, Courtaboeuf, France) according to the manufacturer's recommendations. cDNA synthesis was carried out for 90 min at 42°C in a reaction mixture containing two units of RNase inhibitor (Promega, Charbonnieres, France), three units of avian myeloblastosis virus reverse transcriptase (Promega), 100 ng of specific reverse primers (Sigma-Genosys Ltd, Saint-Quentin Fallavier, France) and 2.5 mM DNTP (Promega), for 1 μg of total RNA. The cDNA samples were stored at −20°C.
Amplification was achieved by adding 2 μl of cDNA to a PCR mixture containing 2.5 U of Taq DNA polymerase (Promega), 2 mM MgCl2, 1 mM DNTP, 100 ng of each primer and PCR buffer (Promega), in a final volume of 50 μl. PCR was run in a thermal cycler (PerkinElmer Cetus 480), for 35 cycles, as follows: a denaturation step (94°C for 1 min), an annealing step (56°C for 45 s) and an elongation step (72°C for 1 min). The primers for IL-4R (Sigma-Genosys Ltd), published elsewhere41 (Table 1), yielded a PCR product of 510 bp. Each reverse-transcribed mRNA was internally controlled with the 28s ribosomal housekeeping gene.42 PCR products were analyzed on 2% agarose gel containing ethidium bromide.
Real-Time Quantitative PCR for STAT-6, GAPDH, Collagen I, III and IV and for α-SMA Evaluation
STAT-6, GAPDH, type I, III and IV collagens, and α-SMA gene expressions in hIHFs were analyzed by RT–PCR using a LightCycler (Roche Diagnostics, Grenoble, France). The reaction was ensured using the DNA Fast Start SYBR Green Kit (Roche Diagnostics), with LightCycler instruments and technology (Roche Diagnostics). PCR amplification was performed in a total volume of 20 μl in glass capillaries containing 20 ng of each primer (Sigma-Genosys Ltd; Table 1), 3 mM MgCl2, 2 μl Light Cycler Fast Start DNA Master SYBR Green (containing 1.25 U Fast StartTaq polymerase, 10 × Taq buffer, 2 mM of each DNTP, 10 × SYBR Green; Roche Diagnostics), and 2 μl of cDNA (previously diluted to 1/10). The PCR amplification protocol consisted in one step of initial denaturation for 10 min at 94°C, followed by 40 cycles: denaturation (95°C for 10 s), annealing for 5 s (at 70°C for GAPDH, 62°C for STAT-6, 54°C for 28 s, 58°C for collagen I or 65°C for collagen III, IV, α-SMA) and extension (72°C for 5 s). The level of mRNA was calculated by normalizing the threshold cycle (CT) of STAT-6, GAPDH, type I, III and IV collagens, or α-SMA to the CT of the housekeeping gene 28s ribosomal RNA, according to the following formula: the average 28 s CT was subtracted from the average type I, III and IV collagens or P<0.01 (P=0.0001) SMA CT, with the result representing ΔCT. This ΔCT is specific and can be compared with the ΔCT of a calibration sample (control hIHFs). The subtraction of control ΔCT from the ΔCT of the fibroblasts being processed is referred to as ΔΔCT. Relative quantification of the expression of STAT-6, GAPDH, type I, III and IV collagens or α-SMA (by comparison with the control) was determined using the value of .49 The primers used during this study had previously been published (Table 1), and were purchased from Sigma-Genosys Ltd. Each reverse-transcribed mRNA was internally controlled with the 28s ribosomal housekeeping gene,42 for which the primers were designed using Oligo6 software (Table 1). PCR products were analyzed for specificity on 2% agarose gel containing ethidium bromide.
Collagen Detection in Supernatant hIHFs by Sircol Collagen Assay
After deactivation for 48 h in a serum-free medium, hIHFs were cultured in medium with serum and treated with various concentrations of IL-4 (1–500 ng/ml). The supernatant was then removed and assayed for soluble collagen using the Sircol collagen assay, according to the manufacturer's protocol (Sigma). This assay uses Sirius Red, an anionic dye with sulfonic acid side chain groups, which reacts with the basic amino acids present in collagen. Briefly, Sircol reagent was added to the medium and gently agitated for 30 min to allow the collagen–dye complex to form. The samples were then centrifuged at 12 000 r.p.m. for 10 min; the collagen–dye complex precipitate was collected and resolubilized in 0.5 M sodium hydroxide. The dye concentration was estimated by spectrophotometry at 540 nm.
Statistical Analysis
Results are expressed as means±s.d. and the differences between groups were tested using one-way ANOVA and the Mann–Whitney test with StatView IV software (Abacus Concepts, Berkeley, CA, USA). Differences with a P-value <0.05 were considered to be statistically significant.
RESULTS
Isolation and Characterization of hIHFs
Human IHFs obtained from normal human liver and cultured initially on plastic in 5% FCS/DMEM appeared to be morphologically activated; they spread progressively, were flattened and showed a typical myofibroblast-like morphology under phase-contrast microscopy (Figure 1Aa and c). By contrast, cells plated on the Matrigel substrate exhibited a quiescent morphology and formed clusters that were occasionally connected by a filamentous network (Figure 1Ab and d). There was also an intense autofluorescence (vitamin A), of most but not all cells, in Matrigel-cultured (Figure 1Af) but not plastic-cultured (Figure 1Ae) hIHFs.

Morphology and expression of myofibroblast markers in hHIFs cultured on different matrices (plastic and Matrigel). (Aa, Ab) Cells were observed using phase-contrast microscopy; (c, d) cells were stained with Harris hematoxylin and observed using optical microscopy; (e, f) cells were observed using UV microscopy. hIHFs on plastic exhibited a typical myofibroblast-like morphology (a, c). By contrast, hIHFs cultured on Matrigel formed ball-like clusters occasionally connected by a filamentous network (b, d). No autofluorescence (vitamin A) was seen in hIHFs cultured on plastic (e), but strong autofluorescence was observed in hIHFs cultured on Matrigel (f) (magnification × 200). (B) Immunohistochemical and immunofluorescence detection of α-SMA and vimentin in hIHFs cultured on plastic. More than 95% of hIHFs cultured on plastic were stained by an anti-αSMA (c, f and i) or an anti-vimentin (b, e and h) antibody, typical of activated hIFHs (magnification × 100). The immunohistochemical staining of α-SMA and vimentin has been done on cells cytocentrifuged on SuperFrost Plus slides (a–c) and on of cells plated on glass coverslips (d–f), and immunofluorescent staining of cells grown on coverslips (g–i). Staining was consistently absent from negative controls (irrelevant antibody; a, d and g).
The expression of α-SMA, a marker for activated HSCs,50 and vimentin, a marker for cells of mesenchymal origin,51 was used to assess the activation and purity of cultured hIHFs, using immunohistochemistry and immunofluorescence. The expression of α-SMA (Figure 1Bc, f and i) and vimentin (Figure 1Bb, e and h) in hIHFs cultured between passages 2 and 6 on plastic was intense (>98% positive cells for both markers), compared to the negative control (Figure 1Ba, d and g).
hIHFs were also characterized using CD90, a fibroblast marker,52, 53, 54 and CD31, a marker of endothelial cells,52 using flow cytometry and immunofluorescence. More that 95% cells were CD90 positive (Figure 2a), whereas only 1% were CD31 positive (Figure 2b), indicating that hIHFs isolated using our method had not been substantially contaminated by other cell types. Similar results were found with immunofluorescence analysis, all hIHFs being stained by anti-CD90 antibody (Figure 2d) whereas no staining was found in negative controls (Figure 2c).

Detection of CD90 and CD31 on hIHFs using flow cytometry (a, b) and immunofluorescence (c, d). Flow cytometry: (a) hIHFs stained with an anti-human CD90: 93% of cells were CD90 positive; (b) fewer than 1% of cells were CD31 positive. Immunofluorescence: (c) hIHFs stained with an anti-human CD90 antibody: all cells were CD90 positive; (d) negative controls (cells stained with an irrelevant antibody).
IL-4 Induced the Activation of Quiescent hIHFs
The reversion of hIHF activation, assessed in terms of the expression of the activation markers α-SMA, vimentin and prolyl 4-hydroxylase, and analyzed by flow cytometry, was obtained after FCS privation or culture on Matrigel (Figure 3a).

(a) Reversion of hIHFs activation. Cells were seeded on plastic, with or without FSC, or on Matrigel. The expression of activation markers (α-SMA, vimentin and prolyl 4-hydroxylase) was analyzed by flow cytometry. A reversal of hIHF activation was demonstrated after FSC privation or culture on Matrigel (*P<0.0001 vs cells cultured on plastic with FSC); (b) effects of IL-4 on the mRNA expression of α-SMA was analyzed by real-time RT-PCR. The expression of α-SMA mRNA in hIHFs cultured on plastic after FSC privation or Matrigel was significantly enhanced after incubation with TGF-β or IL-4 (*P<0.006, **P<0.04, respectively, vs untreated cells). The enhanced expression of α-SMA after IL-4 treatment of hIHFs was suppressed by anti-IL-4 (#P<0.001 vs treated cells with IL-4 alone). (c) Detection of IL-4R on hIHFs was evaluated by RT–PCR analysis. IL-4R mRNA expression was observed in PBMC, HepG2 cells and hIHFs. IL-4R mRNA was not observed in biliary cells. The housekeeping gene ribosomal 28s was used as an internal control. (d) Immunohistochemical detection of IL-4R: (a) negative controls, hIHFs with an irrelevant antibody; (b) hHIFs expressed IL-4R (original magnification × 400).
The effects of IL-4 on hIHFs activation was then investigated by evaluating α-SMA mRNA levels using real-time RT-PCR analysis in hIHFs previously rendered quiescent by incubation for 2 days in serum-free medium or culture on Matrigel substrate. α-SMA mRNA levels increased significantly after incubation with TGF-β or IL-4, at all the concentrations used (Figure 3b). Anti-human IL-4 (50 ng/ml) suppressed the effect of IL-4 (50 ng/ml) on α-SMA mRNA expression (P<0.01 vs cells treated with IL-4 alone; Figure 3b).
IL-4R Expression by hIHFs
The expression of IL-4R was evaluated in hIHFs by RT–PCR analysis and by immunohistochemistry. With RT–PCR analysis, 28s rRNA amplification, used as an internal control, was positive in all samples. The positive controls for IL-4R mRNA were PBMC and HepG2 hepatocyte line cells that, after amplification, led to the expected 510 pb fragment, characteristic of IL-4R α-chain mRNA (Figure 3c). Biliary cells were used as negative controls. IL-4R mRNA was always detected in hIHFs, whether cultured with or without FCS or on Matrigel (Figure 3c). With immunohistochemical staining, some PBMC exhibited intense, characteristic red staining (data not shown). Negative controls (omitting the primary antibody or staining with an irrelevant antibody) were consistently negative (Figure 3d). By contrast, most hIHFs expressed IL-4R (Figure 3d).
STAT-6 siRNA Transfection Efficiency
The transfection efficiency of STAT-6-specific siRNA was controlled by quantitative RT-PCR (Figure 4a) and western blot (Figure 4b). The relative STAT-6 mRNA level, normalized by internal control 28s, was 0.98±0.03 after cell transfection with the negative control (nonspecific siRNA), 0.89±0.1 for GAPDH siRNA-transfected cells and 0.08±0.02 for STAT-6 siRNA-transfected cells (Figure 4a). Compared with nontransfected control cells (no siRNA), nonspecific siRNA-transfected cells (negative control) and GAPDH siRNA-transfected cells (positive control), STAT-6 mRNA expression was knocked down in STAT-6 siRNA-transfected cells by about 92.0, 91.5 and 90.7%, respectively (P<0.0001 in all cases). In the context of this analysis, GAPDH mRNA levels in GAPDH siRNA-transfected cells were significantly lower (0.02±0.03) than those of nonspecific siRNA-transfected cells (0.80±0.06; P=0.0004) and of STAT-6 siRNA-transfected cells (0.72±0.06; P=0.0006).

Transfection efficiency of hIHFs with STAT-6 siRNA and evaluation of STAT-6 activation after IL-4 treatment. The transfection efficiency of STAT-6 siRNA was checked by RT–PCR for STAT-6 mRNA and by western blot for STAT-6 and P-STAT-6. (a) Negative controls were not transfected (no siRNA or nonspecific siRNA-transfected cells), whereas GAPDH siRNA was used as a positive control for transfection and 28s served as an internal control (*P<0.0001, GAPDH siRNA vs no siRNA and **P<0.0001, STAT-6 siRNA vs no siRNA). (b) STAT-6 siRNA induced a downregulation of cellular STAT-6 and P-STAT-6 protein. β-Actin served as an internal control. (c) Detection of STAT-6 activation by flow cytometry: the percentage of P-STAT-6-positive cells was significantly higher in hIHFs treated with IL-4 than in untreated cells. The addition of anti-IL-4 or transfection with STAT-6 siRNA suppressed this phenomenon. Data are means±s.d., *P<0.0001 vs untreated cells and #P<0.001 vs IL-4 treated cells. (d) Detection of STAT-6 activation by immunohistochemistry: unstimulated hIHFs were negative and hIHFs stimulated with IL-4 (50 ng/ml) were strongly positive for P-STAT-6 (original magnification × 400).
hIHFs transiently transfected with STAT-6 siRNA exhibited minimal STAT-6 protein expression (Figure 4b). In addition to STAT-6 inhibition, a marked reduction in P-STAT-6 was also observed in STAT-6 siRNA-transfected cells treated with IL-4 when compared to nontransfected cells (no siRNA) treated with IL-4 (50 ng/ml) (Figure 4b). By contrast, the transfection of hIHFs with GAPDH-siRNA (positive siRNA control) or with no specific siRNA (negative siRNA control) did not alter STAT-6 protein expression (Figure 4b).
IL-4 Activation of STAT-6 In Vitro
The ability of IL-4 to induce STAT-6 activation was analyzed by flow cytometry (Figure 4c) and immunohistochemistry (Figure 4d). IL-4 proved to be a potent activator of STAT-6, with a higher percentage of cells expressing P-STAT-6 among IL-4 (50 ng/ml)-treated cells for 20 min (57.27±8.40%) than among untreated cells (6.80±2.10%, P<0.001; Figure 4c). The incubation of IL-4-treated hIHFs with anti-human IL-4 (50 ng/ml), or transfection with STAT-6 siRNA, suppressed STAT-6 activation (16.2±3.8 and 9.2±2.4%, respectively, vs cells treated with IL-4 alone; P=0.001 and P<0.0001; Figure 4c).
Absence of IL-4 Toxicity on hIHFs at the Concentrations Used
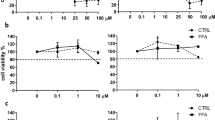
Cells were cultured with C2-Ceramid (20 μM, a hIHFs inductor of apoptosis), or with IL-4 100 ng/ml or TGF-β 10 ng/ml for 48 h (Figure 5). Minimal apoptosis was observed in untreated cells under basal conditions, and reached 7.4±0.7% (percent of Annexin V+/PI− cells) after 48 h of culture, without any difference after the addition of IL-4 or TGF-β, when compared with untreated cells (6.9±0.7 and 11.3±1.4%, respectively). After C2-Ceramid treatment, apoptosis reached 45.4±3.7% (P<0.001 vs untreated cells; Figure 5a). Significant cell necrosis (percent Annexin V+/PI+) was induced by C2-Ceramid (9.6±0.6 vs 1.1±0.1% in untreated cells, P<0.0001), but was not induced by IL-4 or TGF-β treatment (1.0±0.4, 1.2±0.2%, respectively; Figure 5b).

Evaluation of the effects of IL-4 on the apoptosis, necrosis and proliferation of hIHFs. Apoptosis and necrosis were detected by flow cytometry after Annexin V–PI staining. (a) Apoptosis was enhanced after C2-Ceramid treatment of hIHFs, but not after IL-4 treatment. Data are means±s.d., *P<0.0001 vs cells treated with C2-Ceramid. (b) Necrosis was only observed in cells treated with C2-Ceramid and no difference was observed between IL-4- and TGF-β-treated cells and untreated cells (*P<0.001 vs cells treated with C2-Ceramid). (c) Cell proliferation was evaluated by thymidine incorporation. PDGF promoted cell proliferation (positive control), but IL-4 and TGF-β treatment did not alter cell proliferation when compared with untreated cells. Representative results from three different experiments.
Absence of Cell Proliferation Induction by IL-4
The proliferation of hIHFs treated with IL-4, IL-4 plus anti-IL-4, TGF-β or PDGF was assessed by [3H]-thymidine incorporation. The proliferation of hIHFs proved to be significantly stimulated by PDGF, as shown by the increase in [3H]-thymidine incorporation. By contrast, no proliferation was observed in nontreated hIHFs or in cells treated with IL-4 or TGF-β (Figure 5c).
IL-4 Induced Collagen mRNA Expression in hIHFs
TGF-β1, which potently stimulates the expression of procollagen mRNA and other ECM components, was used as a model of a profibrogenic cytokine. The effects of IL-4 and TGF-β1 on the mRNA levels of collagens I, III and IV were investigated in hIHFs rendered quiescent by incubation for 2 days in serum-free medium on plastic, or in hIHFs cultured on Matrigel substrate (Figure 6). TGF-β1 strongly enhanced collagens I, III and IV mRNA levels (14.9±2.9, 26.17±8.2 and 34.2±6.4-fold, respectively; P=0.0004, P=0.0001 and P<0.001 vs untreated cells). Like TGF-β1, IL-4 significantly enhanced collagen I, III and IV mRNA levels in quiescent hIHFs. This effect was dose dependent and was statistically significant as from 10 ng/ml. IL-4 (50 ng/ml) enhanced the mRNA expression of collagens I, III and IV, 14.1±0.2, 16.7±2.4 and 22.4±5.6-fold, respectively, compared to negative controls (P=0.004, P=0.007 and P=0.0005) (Figure 6a–c). When IL-4-treated hIHFs were treated with anti-IL-4 (50 ng/ml) or were transfected with STAT-6 siRNA, the expressions of collagen I, III and IV were reduced to 1.5±0.3 and 2.5±0.1, 2.8±0.4 and 1.8±0.5 and 2.6±0.1 and 5.4±1.4-fold (P=0.01, P=0.01 and P=0.001, respectively, for anti-IL-4, vs cells treated with IL-4 alone and P<0.0001, P=0.001 and P=0.004, respectively, vs cells treated with IL-4 alone, for cells transfected with STAT-6 siRNA).

Evaluation of collagen mRNA expression and collagen production. The effects of IL-4 and TGF-β on collagens I (a), III (b) and IV (c). mRNA levels were investigated on hIHFs rendered quiescent by incubation for 2 days in serum-free medium or culture on Matrigel substrate. IL-4 enhanced collagens I, III and IV mRNA levels in quiescent hIHFs (*P<0.05 on plastic and **P<0.01 on Matrigel vs untreated cells). The enhanced expression of collagen after the IL-4 treatment of hIHFs was suppressed by the addition of anti-IL-4 or STAT-6 siRNA transfection (#P<0.005 vs hIHFs treated with IL-4 alone). (d) Measurement of soluble collagen production in the supernatants of quiescent hIHFs. IL-4 significantly enhanced the secretion of collagen in culture supernatants. This IL-4-induced collagen production was suppressed by the addition of anti-IL-4 or by STAT-6 siRNA transfection (#P<0.005 vs IL-4-treated cells). Representative results from six different experiments.
Basal collagen I, III and IV mRNA levels were low in hIHFs cultured on Matrigel. IL-4 and TGF-β stimulation enhanced these levels (Figure 6a–c): TGF-β increased the expression of collagen I, III and IV that reached 9.2±1.3, 9.6±1.6 and 17.2±3.7-fold, respectively (P<0.001, 0.008 and 0.002 vs untreated cells). IL-4 (50 ng/ml) treatment also increased the mRNA expression of collagens I (9.4±0.7-fold), III (8.9±0.5-fold) and IV (15.2±1.8-fold), compared to negative controls (P<0.0001, P=0.001 and P<0.001, respectively). This effect was dose dependent and was statistically significant as from 10 ng/ml. Anti-IL-4 (100 ng/ml) suppressed this effect, with collagen I, III and IV expressions reduced to 3.2±0.4, 3.3±0.6 and 4.0±0.9-fold (P=0.0002, P=0.02 and P=0.004, respectively, vs cells treated with IL-4 alone). No statistically significant differences in collagen mRNA levels were found between the two groups of quiescent cells. The expressions of collagen I, III and IV, induced by IL-4, increased in a time-dependent manner until 48 h, and then decreased at 72 h (data not shown).
Measurement of Collagen Production in Supernatants
After 48 h of treatment, collagen production was quantified by a Sircol assay in the supernatants of hIHFs treated with IL-4 (5–500 ng/ml) or TGF-β (10 ng/ml). IL-4 (50 ng/ml) significantly increased the production of collagen by 11.3±1.7 and 10.1±1.2 μg/ml, respectively (P=0.0005 and P=0.002; Figure 6d), by comparison with untreated cells (3.2±0.8 μg/ml). For IL-4, this effect was dose dependent and was statistically significant as from 10 ng/ml. Incubation with anti-IL-4 for 48 h, or transfection by STAT-6 siRNA, significantly reduced collagen production in supernatants (4.6±0.5 μg/ml and 3.7±0.1, P=0.005 and P<0.0001, respectively, compared to cells treated with IL-4 alone; Figure 6d). No statistical differences were found regarding collagen production between the two groups of quiescent cells.
DISCUSSION
HCV-related cirrhosis is a common indication for LT, although the recurrence of HCV infection is universal, and characterized by accelerated hepatic fibrosis.55, 56 We have recently shown that IL-4 expression is elevated in severe recurrent hepatitis C,22 and that IL-4 induces apoptosis of human hepatocytes, which might contribute to the progression of severe liver graft damage.57 Nevertheless, the accelerated progression of fibrosis during recurrent hepatitis C prompted us to advance the hypothesis that IL-4 might directly induce liver fibrosis by activating hIHFs and inducing collagen production.
Little has been reported to date concerning the effect of IL-4 on human liver nonparenchymal cells. IL-4 promotes collagen synthesis by LI90 cells (an HSC line33) and by nonparenchymal human liver cells in vitro. Most studies of liver fibrosis reported until now have considered that activated HSC was the principal fibrogenic cell type contributing to ECM accumulation.58 However, other cell types such as portal fibroblasts may, after activation, acquire fibrogenic potential.59, 60 In our study, a heterogeneous population of hIHFs was isolated from human livers using collagenase perfusion, because most (but not all) of these cells store vitamin A in the same way as stellate cells, when quiescent.60 The cells we studied were then characterized by their morphology, purity and specificity. When cultured on plastic wells, quiescent hIHFs were activated and exhibited the characteristics of myofibroblasts or activated HSCs; they were barely autofluorescent, but strongly immunoreactive for α-SMA and vimentin. To evaluate IL-4 activity on hIHFs, these cells were first reversed to quiescence by incubation in a serum-free medium or culture on plates coated with a basal membrane-like substrate (Matrigel). We were able to show that cells were thus partially autofluorescent, with a reduced expression of α-SMA, vimentin and prolyl 4-hydroxylase activation markers. To our knowledge, this study shows for the first time that the subsequent incubation of quiescent hIHFs with IL-4 can directly activate cells toward a myofibroblast phenotype with morphological changes, a loss of vitamin A droplets and strong expression of α-SMA and vimentin.
The IL-4 concentrations used during this study were similar to those employed in previous in vitro studies (10–50 ng/ml).61, 62 In a model of IL-4-induced rat hepatitis, IL-4 serum concentrations reached 1800 pg/ml.63 In HIV–Trypanosoma cruzi coinfection, IL-4 serum levels reached 6200 pg/ml.64 Intrahepatic concentrations were unknown. We thus took considerable care to demonstrate that during our experiments IL-4 at the concentrations employed was not toxic for hIHFs and had no effect on cell proliferation.
Our results show for the first time that hIHFs express IL-4R, both in terms of mRNA and protein. The binding of IL-4 to IL-4R triggers the phosphorylation of JAK-1 and JAK-3,65 leading to activation of the major IL-4 signaling pathway mediated by STAT-6.66 Tyrosine-phosphorylated STAT-6 forms homodimers and translocates to the nucleus, where it binds IL-4-responsive elements and induces gene transcription.67 It has been shown elsewhere that in mouse nonhepatic fibroblasts, IL-4 can induce collagen I production through a mechanism involving STAT-6 and the activation of transcription factors,68 and that the promoter region of collagen type I DNA contains a STAT-6-responsive element.69 Our present results show that after IL-4 interacted with its receptor the STAT-6 signaling pathway was activated in hIHFs, and that STAT-6 phosphorylation was detected after 20 min of incubation with IL-4.
One major finding of this study was that human recombinant IL-4 can promote the activation of quiescent hIHFs, and directly induce collagen production. IL-4 induces the transcription of types I, III and IV collagens, and the production and secretion of collagens in cell supernatants. Interestingly, IL-4 appeared to be nearly as potent as TGF-β (when used at concentrations similar to those previously reported) in stimulating collagen production. This IL-4 activity is mediated by the binding of IL-4R expressed on the surface of hIHFs. The STAT-6 signaling pathway is required to induce IL-4-mediated fibrosis, as the blockade of STAT-6 activation by STAT-6 siRNA and anti-IL-4 antibody inhibited IL-4-mediated cell activation and collagen production. Cells were activated into myofibroblasts, but cell proliferation was not required for collagen production by IL-4.
In conclusion, this study shows that IL-4 exerts a potent profibrotic effect by activating hIHFs and inducing collagen production and secretion, these being major components of the ECM during liver fibrosis. This effect requires IL4-R binding and STAT-6 activation. Thus IL-4, which is overexpressed in severe recurrent hepatitis C, may have a potential role in the accelerated course of HCV infection after LT. These findings may have therapeutic implications: drugs that are able to inhibit STAT-6, or immunosuppressive drugs with anti-IL4 activity, might be beneficial in liver transplant recipients experiencing recurrent HCV infection or diseases with a fibrotic course. High IL-4 producers might represent a subset of liver transplant recipients who warrant more intensive management.
References
Kisseleva T, Brenner DA . Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol 2006;21:84–87.
Iredale JP . Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 2007;117:539–548.
Iredale JP . Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis 2001;21:427–436.
Bataller R . Liver fibrosis. J Clin Invest 2005;115:209–218.
Gressner AM . Proliferation and transformation of cultured liver fat-storing cells perisinusoidal lipocytes under conditions of β-D-xyloside-induced abrogation of proteoglycan synthesis. Exp Mol Pathol 1991;55:143–169.
Sato J, Schorey J, Ploplis VA, et al. The fibrinolytic system in dissemination and matrix protein deposition during a mycobacterium infection. Am J Pathol 2003;163:517–531.
Lotersztajn S, Julien B, Teixeira CF, et al. HEPATIC FIBROSIS: molecular mechanisms and drug targets. Annu Rev Pharmacol Toxicol 2005;45:605–628.
Knittel T, Kobold D, Piscaglia F, et al. Localization of liver myofibroblasts and hepatic stellate cells in normal and diseased rat livers: distinct roles of (myo-)fibroblast subpopulations in hepatic tissue repair. Histochem Cell Biol 1999;112:387–401.
Kinnman N, Francoz C, Barbu V, et al. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab Invest 2003;83:163–173.
Friedman SL . Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000;275:2247–2250.
Nagy P, Schaff Z, Lapis K . Immunohistochemical detection of transforming growth factor-beta 1 in fibrotic liver diseases. Hepatol 1991;14:269–273.
Castilla A, Prieto J, Fausto N . Transforming growth factors beta 1 and alpha in chronic liver disease. Effects of interferon alfa therapy. N Engl J Med 1991;324:933–940.
Shimoda M, Ghobrial RM, Carmody IC, et al. Predictors of survival after liver transplantation for hepatocellular carcinoma associated with Hepatitis C. Liver Transpl 2004;10:1478–1486.
Kornberg A, Kqpper B, Tannapfel A, et al. Impact of mycophenolate mofetil vs azathioprine on early recurrence of hepatitis C after liver transplantation. Int Immunopharmacol 2005;5:107–115.
Feray C . Fibrosis progression after liver transplantation in patients with recurrent hepatitis. J of Hepatol 2004;41:862–863.
Curry MP . Hepatitis B and Hepatitis C viruses in liver transplantation. Transplantation 2004;78:955–963.
Berenguer M, Lopez-Labrador FX, Wright TL . Hepatitis C and liver transplantation. J Hepatol 2001;35:666–678.
Schirren CA, Jung MC, Gerlach JT, et al. Liver-derived hepatitis C virus (HCV)-specific CD41 T cells recognize multiple HCV epitopes and produce interferon gamma. Hepatol 2000;32:597–603.
Zekry A, Bishop GA, Bowen DG, et al. Intrahepatic cytokine profiles associated with posttransplantation hepatitis C virus-related liver injury. Liver Transpl 2002;8:292–301.
Boleslawski E, Conti F, Sanquer S, et al. Defective inhibition of peripheral CD8+ T cell IL-2 production by anti-calcineurin drugs during acute liver allograft rejection. Transplantation 2004;77:1815–1820.
Conti F, Calmus Y, Rouer E, et al. Increased expression and role of interleukin-4 during liver allograft rejection. J Hepatol 1999;30:935–943.
Dharancy S, Podevin P, Aoudjehane L, et al. Elevated interleukin-4 expression in severe recurrent hepatitis C virus after liver transplantation. Transplantation 2007;83:906–911.
Kim J, Cheon IS, Won YJ, et al. IL-4 inhibits cell cycle progression of human umbilical vein endothelial cells by affecting p53, p21Waf1, cyclin D1, and cyclin E expression. Mol Cells 2003;16:92–96.
Vella V, Mineo R, Frasca F, et al. Interleukin-4 stimulates papillary thyroid cancer cell survival: implications in patients with thyroid cancer and concomitant Graves' disease. J Clin Endocrinol Metab 2004;89:2880–2889.
Nelms K, Keegan AD, Zamorano J, et al. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunology 1999;17:701–738.
Sempowski GD, Beckmann MP, Derdak S, et al. Subsets of murine lung fibroblasts express receptors membrane-bound and soluble IL-4: role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J Immunol 1994;152:3606–3614.
Doucet C, Brouty-Boye D, Pottin-Clemenceau C, et al. IL-4 and IL-13 specifically increase adhesion molecule and inflammatory cytokine expression in human lung fibroblasts. Int Immunol 1998;10:1421–1433.
Liu X, Conner H, Kobayashi T, et al. Synergetic effect of interleukin-4 and transforming growth factor-beta1 on type I collagen gel contraction and degradation by HFL-1 cells: implication in tissue remodelling. Chest 2003;123:427–428.
Fujitsua Y, Fukudaa K, Kumagaia BN, et al. IL-4-induced cell proliferation and production of extracellular matrix proteins in human conjunctival fibroblasts. Exp Eye Res 2003;76:107–114.
Ando M, Miyazaki E, Fukami T, et al. Interleukin-4-producing cells in idiopathic pulmonary fibrosis: an immunohistochemical study. Respirology 1999;4:383–391.
Salmon-Ehr V, Serpier H, Nawrocki B, et al. Expression of interleukin-4 in scleroderma skin specimens and scleroderma fibroblast cultures. Potential role in fibrosis. Arch Dermatol 1996;132:802–806.
Tiggelman AM, Boers W, Linthorst C, et al. Collagen synthesis by human liver (myo)fibroblasts in culture: evidence for a regulatory role of IL-1 beta, IL-4, TGFβ and IFN gamma. J Hepatol 1995;23:307–317.
Sugimoto R, Enjoji M, Nakamuta M, et al. Effect of IL-4 and IL-13 on collagen production in cultured LI90 human hepatic stellate cells. Liver Int 2005;25:420–428.
Farah IO, Mola PW, Kariuki TM, et al. Repeated exposure induces periportal fibrosis in Schistosoma mansoni-infected baboons: role of TGF-beta and IL-4. J Immunol 2000;164:5337–5343.
Cheever AW, Williams ME, Wynn TA, et al. Anti-lL-4 treatment of Schistosoma mansoni-infected mice inhibits development of T cells and non-B, non-T cells expressing Th2 cytokines while decreasing egg-induced hepatic fibrosis. J Immunol 1994;153:753–759.
Hillaire S, Boucher E, Calmus Y, et al. Effects of bile acids and cholestasis on major histocompatibility complex class I in human and rat hepatocytes. Gastroenterology 1994;107:781–788.
Win KM, Charlotte F, Mallat A, et al. Mitogenic effect of transforming growth factor-beta 1 on human Ito cells in culture: evidence for mediation by endogenous platelet-derived growth factor. Hepatology 1993;18:137–145.
Conti F, Frappier J, Dharancy S, et al. Interleukin-15 production during liver allograft rejection in humans. Transplantation 2003;76:210–216.
Sohara N, Znoyko I, Levy MT, et al. Reversal of activation of human myofibroblast-like cells by culture on a basement membrane-like substrate. J Hepatol 2002;37:214–221.
Davaille J, Li L, Mallat A, et al. Sphingosine 1-phosphate triggers both apoptotic and survival signals for human hepatic myofibroblasts. J Biol Chem 2002;277:37323–37330.
Van der Velden VHJ, Naber BAE, Wierenga-Wolf AF, et al. Interleukin-4 receptors on human bronchial epithelial cells. An in vivo and vitro analysis of expression and function. Cytokine 1998;10:803–813.
De Leeuw WJ, Slagboom PE, Vijg J . Quantitative comparison of mRNA levels in mammalian tissues: 28 s ribosomal RNA level as an accurate internal control. Nucleic Acids Res 1989;23:10137–10138.
Razzaque MS, Ahmed BS, Foster S, et al. Effects of IL-4 on conjunctival fibroblasts: possible role in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci 2003;44:3417–3423.
Harumiya S, Gibson MA, Koshira Y . Antisense suppression of collagen VI synthesis results in reduced expression of collagen I in normal human osteoblast-like cells. Biosci Biotechnol Biochem 2002;66:2743–2747.
Lam S, Van der Geest RN, Verhagen NA, et al. Secretion of collagen type IV by human renal fibroblasts is increased by high glucose via a TGF-beta-independent pathway. Nephrol Dial Transplant 2004;19:1694–1701.
Sugiyama S, Kugiyama K, Nakamura S, et al. Characterization of smooth muscle-like cells in circulating human peripheral blood. Atherosclerosis 2006;187:351–362.
Dondi E, Rogge L, Lutfalla G, et al. Down-modulation of responses to type I IFN upon T cell activation. J Immunol 2003;170:749–756.
Zhang MS, Zhou YF, Zhang WJ, et al. Apoptosis induced by short hairpin RNA-mediated STAT6 gene silencing in human colon cancer cells. Chin Med J 2006;119:801–808.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta (CT)) method. Methods 2001;25:402–408.
Knittel T, Kobold D, Piscaglia F, et al. Localization of liver myofibroblasts and hepatic stellate cells in normal and diseased rat livers: distinct roles of (myo-)fibroblast subpopulations in hepatic tissue repair. Histochem Cell Bio 1999;112:387–401.
Koenig S, Krause P, Drabent B, et al. The expression of mesenchymal, neural and haematopoietic stem cell markers in adult hepatocytes proliferating in vitro. J Hepatol 2006;44:1115–1124.
Saalbach A, Kraft R, Herrmann K, et al. The monoclonal antibody AS02 recognizes a protein on human fibroblasts being highly homologous to Thy-1. Arch Dermatol Res 1998;290:360–366.
Tang L, Luo QL, Xia QJ, et al. The study of CD90 expressing in orbital fibroblasts of thyroid associated ophthalmopathy. Sichuan Da Xue Xue Bao Yi Xue Ban 2006;37:37879–37881.
Raposio E, Guida C, Baldelli I, et al. Characterization and induction of human pre-adipocytes. Toxicol In Vitro 2007;21:330–334.
Berenguer M . Natural history of recurrent hepatitis C. Liver Transpl 2002;8:14–18.
Neumann UP, Berg T, Bahra M, et al. Fibrosis progression after liver transplantation in patients with recurrent hepatitis C. J Hepatol 2004;41:830–836.
Aoudjehane L, Podevin P, Scatton O, et al. Interleukin-4 induces human hepatocyte apoptosis through a Fas-independent pathway. FASEB J 2007;21:1433–1444.
Gallois C, Habib A, Tao J, et al. Role of NF-kappaB in the antiproliferative effect of endothelin-1 and tumor necrosis factor-alpha in human hepatic stellate cells. Involvement of cyclooxygenase-2. J Biol Chem 1998;273:23183–23190.
Gressner AM, Lahme B, Meurer SK, et al. Variable expression of cystatin C in cultured trans-differentiating rat hepatic stellate cells. World J Gastroenterol 2006;12:731–738.
Guyot C, Lepreux S, Combe C, et al. Hepatic fibrosis and cirrhosis. The (myo)fibroblastic cell subpopulations involved. Int J Biochem Cell Biol 2006;38:135–151.
Jaruga B, Hong F, Sun R . Crucial role of IL4/STAT6 in T cell-mediated hepatitis: up-regulation eotaxin and IL5 and recruiting leukocytes. J Immunol 2003;171:3233–3244.
Ethier MF, Madison JM . IL-4 inhibits calcium transients in bovine rachealis cells by a ryanodine receptor dependent mechanism. FASEB J 2005;20:154–156.
Li B, Sun R, Wei H, et al. Interleukin-15 prevents concanavalin A-induced liver injury in mice via NKT cell-dependent mechanism. Hepatology 2006;43:1211–1219.
Rodrigues DB, Correia D, Marra MD, et al. Cytokine serum levels in patients infected by human immunodeficiency virus with and without Trypanosoma cruzi coinfection. Rev Soc Bras Med Trop 2005;38:483–487.
Sasaki Y, Mita H, Toyota M, et al. Identification of the interleukin 4 receptor alpha gene as a direct target for p73. Cancer Res 2003;63:8145–8152.
Wurster AL, Tanaka T, Grusby MJ . The biology of Stat4 and Stat6. Oncogene 2000;19:2577–2584.
Jiang H, Harris MB, Rothman P . IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunology 2000;105:1063–1070.
McGaha TL, Le M, Kodera T, et al. Molecular mechanisms of interleukin-4-induced up-regulation of type I collagen gene expression in murine fibroblasts. Arthritis Rheum 2003;48:2275–2284.
Buttner C, Skupin A, Riebe EP . Transcriptional activation of the type I collagen genes COL1A1 and COL1A2 in fibroblasts by interleukin-4: analysis of the functional collagen promoter sequences. J Cell Physiol 2004;198:248–258.
Author information
Authors and Affiliations
Corresponding author
Additional information
Lynda Aoudjehane is on a fellowship from the Société Francophone de Transplantation.
Rights and permissions
About this article
Cite this article
Aoudjehane, L., Pissaia, A., Scatton, O. et al. Interleukin-4 induces the activation and collagen production of cultured human intrahepatic fibroblasts via the STAT-6 pathway. Lab Invest 88, 973–985 (2008). https://doi.org/10.1038/labinvest.2008.61
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2008.61
Keywords
This article is cited by
-
B cell depletion treatment decreases CD4+IL4+ and CD4+CD40L+ T cells in patients with systemic sclerosis
Rheumatology International (2019)
-
M2 macrophage-mediated interleukin-4 signalling induces myofibroblast phenotype during the progression of benign prostatic hyperplasia
Cell Death & Disease (2018)
-
Development of an in vitro model to test antifibrotic drugs on primary human liver myofibroblasts
Laboratory Investigation (2016)
-
Role of anti-inflammatory cytokines IL-4 and IL-13 in systemic sclerosis
Inflammation Research (2015)
-
LL-37 secreted by epithelium promotes fibroblast collagen production: a potential mechanism of small airway remodeling in chronic obstructive pulmonary disease
Laboratory Investigation (2014)
