
Abstract
Neurodegeneration develops in association with inflammation and demyelination in multiple sclerosis. Available data suggest that the progressive neuroaxonal loss begins in the earliest stages of the disease and underlies the accumulation of clinical disability. The loss of neurons and their processes is driven by a complex molecular mechanism involving cellular and humoral immune histotoxicity, demyelination, reduced neurotrophic support, metabolic impairment, and altered intracellular processes. Here we survey available data concerning the role of autoreactive immunoglobulins in neurotoxicity. A better understanding of molecular pathways leading to immune-mediated neurodegeneration may have key importance in the successful treatment of the disease.
Similar content being viewed by others


Main
Multiple sclerosis (MS) is an inflammatory demyelinating disorder of the central nervous system (CNS).1 Many studies explored as to how the ignition of autoimmunity occurs, why an immunologically privileged site becomes targeted, what the efferent components of immune attacks are, and how the destruction of myelin and oligodendrocytes leads to the clinical presentation of the disease. A generally accepted view of MS pathogenesis has linked the disease process to myelin-specific, CD4+ T lymphocytes which, upon activation by unknown factors, migrate through the blood–brain barrier (BBB), engage CNS-related antigenic peptides presented by antigen presenting cells, clonally expand, and exert cytotoxic attacks on oligodendrocytes and myelin. This hypothesis has been largely driven by observations from the experimental autoimmune encephalomyelitis (EAE) model that represents a demyelinating CNS disorder mediated by myelin-specific CD4+ T cells.2, 3
However, recent histological analyses4 and in vivo studies by magnetic resonance imaging (MRI) and spectroscopy (MRS)5, 6 emphasize that neurodegeneration develops along with inflammatory demyelination. The pathology affects the entire brain, but with different distributions of inflammation, demyelination, and neurodegeneration in the white and gray matter.7 The neuroaxonal loss is likely to be secondary to inflammation and demyelination, but it begins in the earliest stages of the disease and progresses even after the decline of inflammation. Most importantly, neuroaxonal loss represents the major pathological correlate of clinical disability.5 Further evidence to support the importance of neurodegeneration in MS is obtained from clinical data showing only a partial success of the available disease-modifying drugs (interferons, glatiramer acetate, monoclonal antibodies to antigenic determinants expressed on T lymphocytes), which impede activation and migration of inflammatory cells via the BBB, but have no direct effect on the degenerative processes in CNS.8
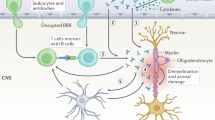
The exploration of neurodegeneration in MS received high priority in the last few years. The data establish that this is a multifactorial process involving loss of myelin protection, immune-mediated histotoxicity, decreased trophic support, mitochondrial damage, metabolic changes, and altered signaling.9, 10, 11, 12 We recently reviewed our and others' published works concerning the involvement of mitochondrial mechanisms in neurodegeneration associated with inflammation.13, 14, 15 As an extension of surveys on neuroaxonal loss related to inflammatory demyelination, the present paper focuses on the involvement of humoral immune factors (Table 1). The available information allows us to generate a preliminary concept of immune-mediated neuronal dysfunction and loss, and to highlight potential pathways amenable to therapeutic interventions (Figure 1; Tables 2, 3 and 4).

Neuroaxonal targets of humoral autoimmunity in MS and EAE. This figure depicts the thus far identified CNS antigens targeted by humoral autoimmunity in inflammatory demyelination.
B CELLS AND HUMORAL AUTOIMMUNITY IN MS
Histological Observations
Varying contributions of humoral immune response to the development of demyelinating lesions have been proposed.61 Type II lesions have prominent IgG and complement-mediated components of myelin destruction in the presence of T lymphocyte and macrophage infiltration, whereas type I lesions are characterized by only cellular elements of inflammation. Signs of autoimmunity are lacking in type III and IV lesions which appear as variants of a primary oligodendrocytopathy. However, humoral autoimmunity is not only involved in certain forms of demyelination, but also instrumental in the process of neurodegeneration but with thus far unknown relative destructivity in various disease subtypes.
Immunologic and Molecular Data
The literature is markedly weighted towards characterization of the cellular arm of autoimmunity, but new data also support that B cells and humoral immunity play important roles in MS. B cells represent about 10–20% of the circulating lymphoid cells and are involved in multiple immune pathways including the presentation of antigenic determinants and the expression of costimulatory signals for T lymphocytes, production of immunoglobulins, secretion of cytokines, and recruitment of T cells into CNS. The B-cell lineage is represented by B lymphocytes (CD19+, CD138−), plasma blasts (CD19+, CD138+), and plasma cells (CD19−, CD138+; for brevity, altogether B cells) in CNS and cerebrospinal fluid (CSF). Most of these cells express memory (CD27+) phenotypes in MS. Therapeutic observations support that B cells may both directly and indirectly contribute to the development of MS. Removal of immunoglobulins from the peripheral blood by plasmapheresis appears to be beneficial in the subgroup of patients with type II lesions59 and depletion of CD20+ B cells by rituximab results in a significant reduction in the number of enhancing MRI lesions. The latter intervention, however, does not exert its beneficial effects by directly affecting the immunoglobulin pool, but by depleting memory B cells and altering antigen presentation, T-cell activation, or T-cell recruitment into the CNS.60
Molecular data also support the involvement of B lymphocytes and their products in MS. Intrathecal production of immunoglobulins with an oligoclonal electrophoretic distribution pattern is a hallmark of the disease. Clonally expanded autoreactive B cells in the CSF and CNS have increased VH mutation rates concentrated in the complementarity determining regions.62 VH sequences expressed in plaques are absent in the peripheral blood.62 Ectopic germinal centers likely contribute to the local generation of B-cell responses in the CNS in both EAE and MS.38, 39 B-cell heavy and light chain editing, a mechanism to prevent autoimmunity by the replacement of elements in rearranged immunoglobulin genes after the re-expression of recombination activating genes—RAG1/RAG2, is inefficient in MS suggested by the detection of autoreactive B cells with unsuccessfully edited receptors in the CSF.63 These observations suggest that CNS antigen-specific and clonally expanded B cells are present in CNS and CSF of MS patients. These B-cell clones exhibit complex molecular characteristics and intraclonal diversity.
Activation of B Cells
What drives B-cell activation and clonal expansion is not fully understood. Epstein–Barr virus (EBV), a B-lymphotropic microorganism, has been implicated in the development of the disease by epidemiological, immune-serological, and histological studies.64, 65 MS patients have intracerebral accumulation of EBV-infected B cells and plasma blasts, mostly in ectopic B-cell follicles,39 and a cross-reactivity of the anti-EBV-specific cells with CNS antigens has been noted.64, 65 Other mechanisms leading to autoimmune B-cell activation may be initiated by CD4+ T helper type 2 (TH2) lymphocytes and their soluble inflammatory products (eg interleukine 4, 5, and 13),66 followed by an antigen-specific expansion of clones either in the peripheral circulation or in the damaged CNS tissue, where intracellular proteins are released. Bystander activation describes a non-antigen-specific activation of T or B cells usually mediated by soluble inflammatory products of nearby immune cells.
Anti-Myelin Antibodies
Immunoglobulins produced by activated B cells in the CNS and CSF target numerous self-antigens including components of the CNS myelin such as myelin basic protein, proteolipid protein, myelin-associated glycoprotein, and myelin oligodendrocyte glycoprotein.67, 68 A great number of papers discusses the involvement of myelin-specific antibodies in the development of demyelination and disease progression.69, 70 Pros and cons for the primary pathogenic significance or the reactive nature of anti-myelin-specific B cells have been debated.71, 72 The use of some of these antibodies as biomarkers to support the diagnosis or to monitor disease activity and course also has been proposed, but not uniformly accepted.73, 74
Anti-Neuronal Antibodies
Autoantibodies that target neuronal molecules and potentially exert histotoxicity have been less scrutinized. Nevertheless, growing data reveal that these antigen–antibody interactions play important roles in a number of neuroinflammatory and neurodegenerative conditions. In contrast to the usual scenario of a T lymphocyte-driven autoimmunity and tissue damage, the humoral immune system appears to be more successful in breaking anti-neuronal tolerance than the cellular system, and has greater pathogenic significance.43 This survey presents information that supports the involvement of the humoral immune response in MS-related neurodegeneration.
ANTI-NEURONAL ANTIBODIES IN MS
Antibodies Targeting Neuronal Cell-Surface Molecules
Neuronal cell-surface antigens
Molecules in the surface membrane of myelinated axons are normally hidden from the immune system, and only become exposed after demyelination when they become antigenic and induce the production of neuron-specific antibodies. IgG and IgM antibodies binding to the surface of a neuronal cell line were found in 70% of sera from patients with secondary progressive (SP)-MS and in 25% of sera from patients with relapsing-remitting (RR)-MS.42 This finding may indicate the spreading of autoimmunity to neuronal antigens as a consequence of CNS tissue damage, and the expansion of pathology from moderate to marked neuroaxonal loss associated with a transition from RR to SP-MS course.
Axolemma-enriched fractions
Antibodies to axolemma-enriched fractions (AEF) of the CNS are also present in CSF and sera of MS patients.16 These antibodies damage neurites and prevent neuronal outgrowth in vitro. The production of anti-axolemma and anti-myelin IgGs appears to be independent. Based on these observations, the use of anti-axolemma IgGs as markers of axonal damage was proposed.55
Neurofascin
One of the targets in antibody-mediated axonal injury is a cell-adhesion molecule neurofascin. The 186 kDa neuron-specific isoform of neurofascin (NF186) is required for the clustering of voltage gated Na+ channels at the nodes of Ranvier. The 155 kDa glial-specific isoform (NF155) is required for the proper assembly of paranodal junction, an important site of interactions between the myelin and axon. Early changes in the distribution of NF155 were observed in MS lesions preceding demyelination.44 Maier et al45 showed that the NF155-levels were reduced and a 40 kDa NF155-fragment increased in plaques, suggesting that NF155 is subject to protein degradation in the lesions.
Levels of antibodies to NF155 and NF186 were significantly higher in sera of patients with chronic progressive forms of MS compared to that of patients with other inflammatory neurological diseases.17 In vitro studies showed that antibodies to neurofascin inhibit axonal conduction. In vivo experiments revealed that antibodies to neurofascin and complement can selectively target nodes of Ranvier, cause axonal injury, and trigger disease exacerbation in EAE. Mathey et al17 also showed that NF155-specific antibodies cross-react with NF186-transfected cells. The recognition of NF186 at the nodes of Ranvier by these antibodies may initiate axonal injury and accelerate disease progression. Neurofascin-specific antibodies can also inhibit remyelination by binding to NF155 expressed on the surface of oligodendrocytes.
Gangliosides
Another group of neuronal antigens includes the gangliosides, which are glycolipids with sialic residues in the outer layer of cell membranes. Gangliosides are particularly enriched in the membranes of neuron. To generate antibody responses, gangliosides do not require major histocompatibility complex (MHC) molecules and T-cell help.75 Although it is unclear whether anti-ganglioside antibodies can cause or result from axonal damage,18 they may certainly serve as a marker of this process. Experimental data reveal that anti-ganglioside antibodies can disrupt the BBB,19 create neuromuscular block by binding to neuronal gangliosides in the neuromuscular junction,56 and inhibit axonal regeneration after peripheral nerve injury in mice.20 Complexes of anti-ganglioside antibodies and complement destroy Schwann cells and myelin membranes in demyelinating neuropathies, and contribute to axonal degeneration in acute motor axonal neuropathy.41 Increased levels of anti-GM3 (monosialo-ganglioside) antibodies can be found in sera of a great proportion of patients with progressive forms of MS (56.3% in primary progressive (PP)-MS and 42.9% in SP-MS vs 2.9% in RR-MS and 14.6% in OND).18 Anti-GD2 (disialo-ganglioside)-like IgM autoantibodies were detected in sera of 30% of MS patients, and a positive correlation of anti-GD2-like IgM reactivity with neurological disability was observed.21 The increased prevalence of GD2-specific IgM antibodies in SP-MS (47.8%) compared to RR-MS (24.2%) and PP-MS (26.7%) also suggests the involvement of these antibodies in inflammation-induced neurodegeneration.
In summary, these data suggest that antibodies specific to neuronal cell-surface molecules are produced during demyelination and may themselves contribute to axonal injury in MS. These antibodies can activate complement and exert cytotoxicity, provide binding sites for the Fc receptors on macrophages and microglial cells, interrupt axon–myelin interaction, inhibit axonal conduction and outgrowth, disrupt the BBB, and alter oligodendrocyte functioning. Correlation of the antibody titers with the severity of disability offers an opportunity of using these neuronal cell-surface antibodies as biomarkers.
Antibodies to Cytoskeletal Proteins
Neurofilaments
Neurofilaments are a group of cytoskeletal proteins expressed in neuronal cells and axons. A variety of neurodegenerative disorders like Alzheimer's disease, Parkinson's disease, and dementia with Lewy bodies are characterized by accumulation of NF proteins signifying abnormalities in the axonal transport and an impending neuronal loss.76, 77
Antibodies to axonal cytoskeletal proteins may be markers of axonal damage, as well as important contributors to neurodegeneration and clinical disability in MS. CSF levels of antibodies against the light subunit of neurofilaments (NFL) correlate with the disease duration, clinical disability, IgG index, and the degree of cerebral atrophy measured by MRI.22, 23 Although elevated levels of NFL-specific antibodies are present in sera of patients with PP-MS,24 these antibodies are also increased in sera of patients with OND.22
Bartos et al40 detected increased intrathecal IgG and IgM antibodies against the medium subunit of neurofilaments (NFM) in patients with all subtypes of MS. Unexpectedly, however, anti-NFM antibody levels appeared to be higher in the serum than in CSF, possibly related to NF antigen leakage from the CNS to the peripheral blood, or to the higher concentration of plasma cells in the blood. Alternatively, anti-NFM antibodies may be triggered by exogenous antigens and molecular mimicry in the peripheral blood.
Although no human data are available to define if anti-neurofilament antibodies themselves contribute to further neuroaxonal damage, experimental models provide supportive evidence. Immunization of mice with NFL triggers the development of a CNS disease characterized by axonal damage, paralysis, and spasticity.25 Depositions of immunoglobulins is seen in the axons of diseased but not of asymptomatic animals. Endocytosis via clathrin-associated vesicles after binding to cell-surface Fc receptors is believed to be the mechanism of getting immunoglobulins into neurons in this model system as well as in MS. Further, microinjections of anti-kinesin or anti-dynein antibodies cause impairment of the anterograde and retrograde NF transport in cultured neurons26 and induce the formation of long and branched mitochondrial structures redistributed to the nuclear periphery.46 These intracellular changes are associated with altered calcium homeostasis,47 apoptosis, and neurodegeneration.48
In summary, these observations suggest that antibodies to neurofilaments and possibly to other cytoskeletal proteins are produced during tissue damage in MS and OND. Experimental data support that these antibodies may get access to their intracellular target and cause changes in axonal transport, mitochondrial distribution and calcium homeostasis, and thus contribute to apoptosis and neurodegeneration.
Antibodies to Various Intracellular Molecules
Forooghian et al78 showed that MS patients have a greater prevalence of T cells specific for several intraneuronal antigens such as neuron-specific enolase (metabolic enzyme), β-arrestin, and retinal arrestin than healthy controls.
Arrestins
Arrestins are a family of multifunctional, intracellular proteins that play important roles in regulating signal transduction and the activity of G-protein-coupled receptors.
Anti-β-arrestin-specific antibodies and antibodies to retinal arrestin can be found in sera of patients with MS.27, 28 β-arrestin-1 enhances the expression of antiapoptotic Bcl2 that may control the development of both MS and EAE. β-Arrestin-1-knockout mice are more resistant, and β-arrestin-1 transgenic mice are more susceptible to EAE.57 These data suggest that pharmacologic downregulation of β-arrestin-1 expression may represent a reasonable approach in MS.58
GAD
Immunity to glutamate decarboxylase (GAD), an enzyme that converts glutamate into the inhibitory neurotransmitter aminobutyric acid (GABA), can also be detected in MS. GAD is expressed in various cell types including neurons,29 and its activity is reduced in sera of MS patients.79 Serum anti-GAD65 antibodies are present in 10% of MS patients.29 However, anti-GAD antibodies are also associated with a number of autoimmune disorders, such as stiff-person syndrome, type 1 diabetes, and Batten disease, and their pathogenic significance remains to be determined.49, 50
HSP
Heat shock proteins (HSPs) are molecular chaperones with increased expression during exposure to elevated temperature or stress. They are implicated in several autoimmune diseases such as autoimmune arthritis, type 1 diabetes mellitus, atherosclerosis, and MS.80 The concentration of anti-α-crystalline antibodies in sera of MS patients correlates with activity as well as severity of the disease.30 In CSF, IgG antibodies to small HSPs (eg HSP27, λA crystallin and λB crystalline) are not increased, but to HSP70 and HSC70 (heat shock cognate protein 70) are elevated and correlate with the disease course.31 Increased antibody titers to HSP60 are present in CSF of both patients with MS and with OND.32 However, no elevated titers of anti-HSP90β antibodies are seen in CSF of normal or OND controls, whereas these immunoglobulins are detected in MS patients during both relapses and remissions.51
Nuclear antigens
Elevated antinuclear antibodies (ANA) in sera of MS patients have been reported at varying frequencies (2.5–81%) depending of methodological approaches.33, 34 These antibodies can be pathognostic for SLE with titers usually reaching 1:640 or higher. In contrast, the ANA titers are low (between 1:40 and 1:100) in sera of MS patients,33, 35 who also often have low-affinity IgG antibodies to multiple other nuclear and cytoplasmic epitopes.35 However, IgG molecules against phosphorylated apoptosis-related proteins detected in SLE patients are completely absent from both the sera and CSF of MS patients. These data suggest that detection of ANA and related antibodies in MS may result from a nonspecific immune activation.
In summary, antibodies to a variety of intracellular molecules including enzymes, signaling molecules, HSPs, and nuclear proteins are detected in MS and other inflammatory and neurological disorders. The production of these antibodies may be related to bystander immune activation and epitope spreading during tissue injury. The pathogenic significance of most of these antibodies remains uncertain.
NEUROTOXICITY AND AUTOIMMUNITY IN NEURODEGENERATIVE DISORDERS
Glutamate-Mediated Neurotoxicity and Autoimmunity
Glutamate
Glutamate is a neurotransmitter released by neurons into the synaptic space where it binds to its postsynaptic receptors. Elevated levels of extracellular glutamate can lead to the death of neurons, astrocytes, and oligodendrocytes.81 Excitotoxic tissue damage mediated by glutamate has been described in a number of neurologic diseases (eg stroke, traumatic injury, neurodegeneration) including MS.82
Glutamate Transporters
Glial cells and neurons express various types of glutamate excitatory amino-acid transporters (EAAT1–EAAT5). The re-uptake of glutamate appears to be impaired in MS due to the downregulation of EAAT1 and EAAT2 molecules in both white matter83 and cortical lesions.84 MRS studies also reveal increased amounts of glutamate in the normal-appearing white matter and acute contrast-enhancing lesions of MS.85 Glutamate levels in the CSF are higher during relapses than remissions, and correlate with the disease severity.86, 87 These observations suggest that inflammation upsets the balance of glutamate release and re-uptake, and the excessive glutamate may escalate the tissue injury in MS.
Glutamate receptors
Glutamate toxicity may be further enhanced by altered receptor expression and signaling.88 Two main subtypes of glutamate receptors have been identified: ionotropic, coupled directly to membrane ion channels; metabotropic, coupled to G proteins. The ionotropic receptors are divided into three further subtypes based on their selective agonists: N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), and kainite.82 Newcombe et al89 reported an elevated expression of subunit 1 of AMPA receptor (GluR1) on oligodendrocytes at the borders of active plaques, and subunit 3 of AMPA receptor (GluR3) and metabotropic glutamate receptors (mGluR) on reactive astrocytes in MS lesions. Activated microglia and macrophages are immunopositive for NMDA receptor subunit 1 (NR1) in plaques89 and may also play a role in Ca2+-dependent injury of oligodendrocytes and neurons.90 NMDA receptor antagonists, memantine, amantadine, and MK-801 reduce neurological deficits in EAE,91, 92 and blockade of the AMPA receptors by antagonists also ameliorate clinical signs of EAE.93, 94
Antibodies targeting glutamate receptors may have agonist or antagonist effects. Agonists usually cause excitotoxicity and complement-mediated cell death.95, 96 Anti-GluR3 antibodies have been implicated in various epilepsy syndromes (noninflammatory focal epilepsy, ‘catastrophic’ epilepsy, Rasmussen's encephalitis), but their causative involvement in neuronal damage remains controversial.97, 98, 99 Although such antibodies have not been reported in MS, their presence and pathogenic contribution to impaired glutamate homeostasis and signaling cannot be excluded until systematically tested.
In summary, these observations suggest that glutamate homeostasis is being upset in inflammatory lesions of MS, where the concentration of glutamate is increased, at least in part, due to a decreased re-uptake. In addition, the expression of glutamate receptors is altered, which can impact on intracellular signaling. Anti-glutamate receptor antibodies are associated with inflammatory and neurodegenerative disorders, and their clearance often correlate with clinical improvement.
Antibodies in Paraneoplastic Neurological Syndromes and MS
Paraneoplastic neurological syndromes (PNSs) are mediated by immune reactions against autoantigens shared between cancer cells and self-antigens, most commonly expressed in the peripheral or CNS. Paraneoplastic antibodies are associated with various forms of neurodegeneration in PNSs,100 and occasionally also can be detected in MS. However, the most common paraneoplastic immunoglobulins (eg anti-Hu, anti-Yo, and anti-Ri) have not been reported in MS.
NMDA receptor
Although NMDA-receptor-specific antibodies have not been reported in sera or CSF of MS patients, a role for these antibodies is suggested in other inflammatory CNS disorders. A newer form of encephalitis in women with or without ovarian teratomas is associated with antibodies against the NR1/NR2 subunits of the NMDA receptor.101, 102 As these antibodies disappear upon clinical improvement, their pathogenic involvement is likely in both the paraneoplastic and the autoimmune forms of this encephalitis.
Proteosome
Antibodies to proteasome, a ubiquitous protease complex composed of 14 different subunits, have been detected in paraneoplastic cerebellar degeneration and MS.37, 103 The proteasome complex is involved in protein degradation, processing of transcription factors in apoptosis and the generation of antigenic peptides ultimately presented by the MHC class I molecule.104 Specific autoantibodies against proteasome subunits C2, C8, C9, and C5 were found in sera and CSF of patients with all forms of MS.37 B- and T-cell autoreactivity against the proteasome develops not only in MS, but also in systemic autoimmune diseases and sarcoidosis. However, the prevalence of anti-proteasome antibodies is significantly higher in MS.37
hnRNPs
Heterogeneous nuclear ribonucleoproteins (hnRNPs) play important roles as autoantigens in both autoimmune disorders and PNSs. Antibodies against hnRNP protein B1 are present in CSF but not in sera of most MS patients, and absent in sera or CSF of OND patients.36 The hnRNPs are the major component of nuclear core complex in mammalian cells. The hnRNP A2/B1 proteins provide binding sites for the vaccinia virus.105 Infection of the CNS by a virus (eg EBV or vaccinia) may trigger cross-reactivity with hnRNP A2/B1.36 Alternatively, the damage of oligodendrocytes may result in a release of hnRNP A2/B1 proteins inducing autoimmune response and epitope spreading in MS.
CRMP5
Ducray et al106 showed that serum of a patient with paraneoplastic encephalitis presenting with a Devic syndrome-like phenotype carries anti-CV2/collapsin response-mediated protein-5 (CRMP5) antibodies. Anti-CV2 antibodies react with the developmentally regulated neural proteins CRMPs, particularly with CRMP5.107 Cross et al108 recently reported three patients who had myelopathy with optic neuritis, anti-CV2/CRMP5 antibodies, and cancer. Although both optic neuritis and myelitis can be the presentations of either paraneoplastic syndrome or MS, these observations underscore the role of these antibodies in overlapping syndromes and the importance of a thorough differential diagnosis.
In summary, there is occasionally a phenotypic and immunological overlap between paraneoplastic syndromes and MS. Although the differential diagnosis is usually not very challenging, it is important to be aware that the antibodies characteristically detected in PNSs may also be present in sera of MS patients. In MS, the trigger for the production of these immunoglobulins is, however, different and likely signifies epitope spreading and bystander immune activation.
Anti-Neuronal Antibodies in Patients with Leber Hereditary Optic Nerve Atrophy
Tubulin
An association between Leber hereditary optic nerve atrophy (LHON) and MS was observed long time ago,109 and confirmed when the detection of primary pathogenic LHON mutations at mitochondrial (mt)DNA nucleotides 3460, 11 778, and 14 484 became available. In addition to the detection of MS-like MRI lesions in patients with LHON type of optic nerve atrophy,110 pathogenic LHON mutations were also detected more often than expected by chance in the MS population.13 Therefore, the question arose if these mtDNA mutations trigger autoimmunity leading to inflammatory demyelination and neurodegeneration. The autoimmune hypothesis seemed to gain support, when increased amounts of serum antibodies to tubulin (a human optic nerve globular protein in microtubules) were found in a significant proportion of patients with LHON.111 The absence of anti-tubulin antibodies in nongenetic optic neuropathies suggests that the finding in patients with LHON is not simply due to a nonspecific immune response to damaged optic nerve proteins.111
In summary, anti-tubulin antibodies may play a role in the development of inflammatory demyelination and neurodegeneration in patients carrying LHON mtDNA mutations.
PROTECTIVE ANTIBODIES IN MS
Autoreactive antibodies cannot only be destructive but also can be protective. Monoclonal IgM antibodies to CNS glial cells can promote remyelination in murine models, and autoreactive IgM antibodies represent an endogenous system involved in physiologic mechanisms of tissue repair.52, 53 These autoreactive IgM antibodies recognize a variety of proteins on the surface of not only glial cells but also other cell types including neurons. Polyspecific autoreactive IgM antibodies are present in sera of MS patients and are likely involved in the activation of complements needed for an efficient phagocytosis and removal of myelin debris by macrophages in demyelinating lesions.54 In addition to clearing debris, IgM antibodies may also promote remyelination by binding to the surface of oligodendrocytes and induction of intracellular signaling. The role of calcium in the regulation of oligodendrocyte function has been emphasized, because calcium fluxes were observed in astrocytes and oligodendrocytes after treatment of glial cultures with remyelination-promoting antibodies.52, 53 It was also shown that remyelination induced by the human remyelination-promoting antibody rHIgM22 is independent of immunomodulation. This antibody did not specifically bind to any immune cell type derived from the spleen, did not influence the humoral immune response to a T-cell dependent antigen (ovalbumin), and did not alter antigen-specific proliferation of CD8+ and CD4+ cells or cytokine production.112
In summary, these observations suggest that the humoral immune system may act as a double-edged sword both promoting damage and repair. Low-affinity autoreactive antibodies are part of the normal immunoglobulin repertoire in healthy individuals. These antibodies gain major physiologic importance during tissue injury when they contribute to the clearance of damage and promote repair. This endogenous mechanism offers a therapeutic paradigm that has not been exhausted in humans.
CONCLUSION: THE ROLE OF ANTI-NEURONAL ANTIBODIES IN MS
The prevailing view defines MS as a demyelinating neurodegenerative disorder of CNS, which is primarily mediated by activated CNS antigen-specific T cells with a well-orchestrated involvement of various antigen-presenting cells and immunoglobulin-producing B cells. The cause of autoimmunity remains unknown, but an initial activation of CNS antigen-specific T- and B cells by environmental factors possibly involving molecular mimicry, and a secondary activation involving tissue injury, autoantigen release, epitope spreading, and bystander mechanisms during the course of the disease have been considered. As it is not the extent of myelin injury but the extent of neuroaxonal loss that best correlates with clinical disability, the exploration of MS-related neurodegeneration has great practical importance. Although our understanding of this process is incomplete, the available data reveal that neuroaxonal loss is related to a complex mechanism with multiple upstream elements. This survey is focused on what is presently known about the generation and pathogenic significance of CNS antigen-specific immunoglobulins in inflammation-related neurodegeneration (Figure 1; Tables 2 and 3). Although numerous cell-surface and intracellular proteins have been identified as targets to the cellular and humoral immune response, the above list of neuronal autoantibodies is expected to grow rapidly with the application of newer technologies. However, it has already been established that most of the anti-neuronal antibodies (eg anti-AEF, anti-NFL, and anti-ganglioside antibodies) correlate with CNS injury, but are not necessarily specific for MS. Proving the pathogenicity of these antibodies is often difficult in humans, and only data from in vitro studies or experimental models provide evidence to support their biological effects (eg antibodies to neurofascin, gangliosides, NFL, kinesin, and dynein; β-arrestin-1 transgenic; and knockout mice). Questions regarding the endocytosis of autoantibodies and consequences of intracellular antigen-antibody engagement in neurons remain unanswered. As the concentration of these antibodies is likely higher in the CNS than in the serum or even in the CSF, the removal of autoreactive immunoglobulins from the circulation by plasma exchange has limited utility in MS. Elimination of CD20+ B cells by rituximab is although very promising, the clinical benefits of this intervention are likely related to a suppression of T-cell activation rather than an immediate suppression of immunoglobulin production. The endogenous pool of protective autoantibodies has not been exploited in humans, but development of vaccination methods to boost this repertoire is underway (Table 4). Taking all into consideration, the most efficient current treatment modality for neurodegeneration remains an early and aggressive anti-inflammatory intervention, because the prevention of tissue injury may best control the escalating T-cell driven and bystander B-cell activation, continuing BBB break-down and epitope spreading, which can all perpetuate neuroaxonal injury.
References
Bruck W, Stadelmann C . The spectrum of multiple sclerosis: new lessons from pathology. Curr Opin Neurol 2005;18:221–224.
Chitnis T . The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol 2007;79:43–72.
Bynoe MS, Bonorino P, Viret C . Control of experimental autoimmune encephalomyelitis by CD4+ suppressor T cells: peripheral versus in situ immunoregulation. J Neuroimmunol 2007;191:61–69.
Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278–285.
Traboulsee A . MRI relapses have significant pathologic and clinical implications in multiple sclerosis. J Neurol Sci 2007;256 (Suppl 1):S19–S22.
Zivadinov R, Cox JL . Neuroimaging in multiple sclerosis. Int Rev Neurobiol 2007;79:449–474.
Peterson JW, Bö L, Mörk S, et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 2001;50:389–400.
De Jager PL, Hafler DA . New therapeutic approaches for multiple sclerosis. Annu Rev Med 2007;58:417–432.
Sobel RA . The extracellular matrix in multiple sclerosis lesions. J Neuropathol Exp Neurol 1998;57:205–217.
Klawiter EC, Cross AH . B cells: no longer the nondominant arm of multiple sclerosis. Curr Neurol Neurosci Rep 2007;7:231–238.
Lindberg RL, Achtnichts L, Hoffmann F, et al. Natalizumab alters transcriptional expression profiles of blood cell subpopulations of multiple sclerosis patients. J Neuroimmunol 2008;194:153–164.
Ghafourifar P, Mousavizadeh K, Parihar MS, et al. Mitochondria in multiple sclerosis. Front Biosci 2008;13:3116–3126.
Kalman B . Role of mitochondria in multiple sclerosis. Curr Neurol Neurosci Rep 2006;6:244–252.
Kalman B, Laitinen K, Komoly S . The involvement of mitochondria in the pathogenesis of multiple sclerosis. J Neuroimmunol 2007;188:1–12.
Blokhin A, Vyshkina T, Komoly S, et al. Lack of mitochondrial DNA deletions in lesions of multiple sclerosis. Neuromolecular Med 2008;Feb:20;Epub.
DeVries GH . Cryptic axonal antigens and axonal loss in multiple sclerosis. Neurochem Res 2004;29:1999–2006.
Mathey EK, Derfuss T, Storch MK, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med 2007;204:2363–2372.
Sadatipour BT, Greer JM, Pender MP . Increased circulating antiganglioside antibodies in primary and secondary progressive multiple sclerosis. Ann Neurol 1998;44:980–983.
Kanda T, Iwasaki T, Yamawaki M, et al. Anti-GM1 antibody facilitates leakage in an in vitro blood-nerve barrier model. Neurology 2000;55:585–587.
Lehmann HC, Lopez PH, Zhang G, et al. Passive immunization with anti-ganglioside antibodies directly inhibits axon regeneration in an animal model. J Neurosci 2007;27:27–34.
Marconi S, Acler M, Lovato L, et al. Anti-GD2-like IgM autoreactivity in multiple sclerosis patients. Mult Scler 2006;12:302–308.
Eikelenboom MJ, Petzold A, Lazeron RH, et al. Multiple sclerosis: neurofilament light chain antibodies are correlated to cerebral atrophy. Neurology 2003;60:219–223.
Bartos A, Fialová L, Soukupová J, et al. Antibodies against light neurofilaments in multiple sclerosis patients. Acta Neurol Scand 2007a;116:100–107.
Ehling R, Lutterotti A, Wanschitz J, et al. Increased frequencies of serum antibodies to neurofilament light in patients with primary chronic progressive multiple sclerosis. Mult Scler 2004;10:601–606.
Huizinga R, Heijmans N, Schubert P, et al. Immunization with neurofilament light protein induces spastic paresis and axonal degeneration in Biozzi ABH mice. J Neuropathol Exp Neurol 2007;66:295–304.
Theiss C, Napirei M, Meller K . Impairment of anterograde and retrograde neurofilament transport after anti-kinesin and anti-dynein antibody microinjection in chicken dorsal root ganglia. Eur J Cell Biol 2005;84:29–43.
Sudo A, Endo M, Saitoh S . Serum anti-arrestin antibody and disease activity of multiple sclerosis—a case report of 4-year-old child. No To Hattatsu 2000;32:415–419.
Gorczyca WA, Ejma M, Witkowska D, et al. Retinal antigens are recognized by antibodies present in sera of patients with multiple sclerosis. Ophthalmic Res 2004;36:120–123.
Hermitte L, Martin-Moutot N, Boucraut J, et al. Humoral immunity against glutamic acid decarboxylase and tyrosine phosphatase IA-2. J Clin Immunol 2000;20:287–293.
Agius MA, Kirvan CA, Schafer AL, et al. High prevalence of anti-alpha-crystallin antibodies in multiple sclerosis: correlation with severity and activity of disease. Acta Neurol Scand 1999;100:139–147.
Chiba S, Yokota S, Yonekura K, et al. Autoantibodies against HSP70 family proteins were detected in the cerebrospinal fluid from patients with multiple sclerosis. J Neurol Sci 2006;241:39–43.
Yonekura K, Yokota S, Tanaka S, et al. Prevalence of anti-heat shock protein antibodies in cerebrospinal fluids of patients with Guillain-Barré syndrome. J Neuroimmunol 2004;156:204–209.
Roussel V, Yi F, Jauberteau MO, et al. Prevalence and clinical significance of anti-phospholipid antibodies in multiple sclerosis: a study of 89 patients. J Autoimmun 2000;14:259–265.
Ferreira S, D'Cruz DP, Hughes GRV . Multiple sclerosis, neuropsychiatric lupus and antiphospholipid syndrome: where do we stand? Rheumatology (Oxford) 2005;44:434–442.
Lu F, Kalman B . Autoreactive IgG to intracellular proteins in sera of MS patients. J Neuroimmunol 1999;99:72–81.
Sueoka E, Yukitake M, Iwanaga K, et al. Autoantibodies against heterogeneous nuclear ribonucleoprotein B1 in CSF of MS patients. Ann Neurol 2004;56:778–786.
Mayo I, Arribas J, Villoslada P, et al. The proteasome is a major autoantigen in multiple sclerosis. Brain 2002;125:2658–2667.
Cepok S, Rosche B, Grummel V, et al. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain 2005;128 (Part 7):1667–1676.
Serafini B, Rosicarelli B, Franciotta D, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med 2007;204:2899–2912.
Bartos A, Fialová L, Soukupová J, et al. Elevated intrathecal antibodies against the medium neurofilament subunit in multiple sclerosis. J Neurol 2007b;254:20–25.
Willison HJ . Basic and clinical aspects of autoimmune disorders in peripheral nerves. Acta Neurol Scand Suppl 2006;183:14–18.
Lily O, Palace J, Vincent A . Serum autoantibodies to cell surface determinants in multiple sclerosis: a flow cytometric study. Brain 2004;127 (Part 2):269–279.
Huizinga R, Linington C, Amor S . Resistance is futile: antineuronal autoimmunity in multiple sclerosis. Trends Immunol 2008;29:54–60.
Howell OW, Palser A, Polito A, et al. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain 2006;129 (Part 12):3173–3185.
Maier O, Baron W, Hoekstra D . Reduced raft-association of NF155 in active MS-lesions is accompanied by the disruption of the paranodal junction. Glia 2007;55:885–895.
Varadi A, Johnson-Cadwell LI, Cirulli V, et al. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J Cell Sci 2004;117 (Part 19):4389–4400.
Rutter GA, Rizzuto R . Regulation of mitochondrial metabolism by ER Ca2+ release: an intimate connection. Trends Biochem Sci 2000;25:215–221.
Karbowski M, Youle RJ . Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ 2003;10:870–880.
Espay AJ, Chen R . Rigidity and spasms from autoimmune encephalomyelopathies: stiff-person syndrome. Muscle Nerve 2006;34:677–690.
Taplin CE, Barker JM . Autoantibodies in type 1 diabetes. Autoimmunity 2008;41:11–18.
Cid C, Regidor I, Alcázar A . Anti-heat shock protein 90beta antibodies are detected in patients with multiple sclerosis during remission. J Neuroimmunol 2007;184:223–226.
Bieber AJ, Warrington A, Pease LR, et al. Humoral autoimmunity as a mediator of CNS repair. Trends Neurosci 2001;24:S39–S44.
Soldan MM, Warrington AE, Bieber AJ, et al. Remyelination-promoting antibodies activate distinct Ca(2+) influx pathways in astrocytes and oligodendrocytes: relationship to the mechanism of myelin repair. Mol Cell Neurosci 2003;22:14–24.
DeJong BA, Smith ME . A role for complement in phagocytosis of myelin. Neurochem 1997;22:491–498.
Zaffaroni M . Biological indicators of the neurodegenerative phase of multiple sclerosis. Neurol Sci 2003;24 (Suppl 5):S279–S282.
Winer JB . Guillain Barre syndrome. Mol Pathol 2001;54:381–385.
Shi Y, Feng Y, Kang J, et al. Critical regulation of CD4+ T cell survival and autoimmunity by beta-arrestin-1. Nat Immunol 2007;8:817–824.
Frederick TJ, Miller SD . Arresting autoimmunity by blocking beta-arrestin-1. Nat Immunol 2007;8:791–792.
Keegan M, König F, McClelland R, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 2005;366:579–582.
Hauser SL, Waubant E, Arnold DL, et al., HERMES Trial Group. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358:676–688.
Lucchinetti C, Brück W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–717.
Owens GP, Burgoon MP, Anthony J, et al. The immunoglobulin G heavy chain repertoire in multiple sclerosis plaques is distinct from the heavy chain repertoire in peripheral blood lymphocytes. Clin Immunol 2001;98:258–263.
Lambracht-Washington D, O'Connor KC, Cameron EM, et al. Antigen specificity of clonally expanded and receptor edited cerebrospinal fluid B cells from patients with relapsing remitting MS. J Neuroimmunol 2007;186:164–176.
Ascherio A, Munger KL . Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol 2007;61:288–299.
Banwell B, Krupp L, Kennedy J, et al. Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol 2007;6:773–781.
Dhib-Jalbut S . Pathogenesis of myelin/oligodendrocyte damage in multiple sclerosis. Neurology 2007;68 (Suppl 3):S13–S21.
Genain CP, Cannella B, Hauser SL, et al. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med 1999;5:170–175.
O'Connor KC, Appel H, Bregoli L, et al. Antibodies from inflamed central nervous system tissue recognize myelin oligodendrocyte glycoprotein. J Immunol 2005;175:1974–1982.
Dutta R, Trapp BD . Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology 2007;68 (Suppl 3):S22–S31.
Vaknin-Dembinsky A, Weiner HL . Relationship of immunologic abnormalities and disease stage in multiple sclerosis: implications for therapy. J Neurol Sci 2007;259:90–94.
Holmoy T . Immunopathogenesis of multiple sclerosis: concepts and controversies. Acta Neurol Scand Suppl 2007;187:39–45.
Pender MP, Greer JM . Immunology of multiple sclerosis. Curr Allergy Asthma Rep 2007;7:285–292.
Amor S, Giovannoni G . Antibodies to myelin oligodendrocyte glycoprotein as a biomarker in multiple sclerosis—are we there yet? Mult Scler 2007;13:1083–1085.
Lutterotti A, Berger T, Reindl M . Biological markers for multiple sclerosis. Curr Med Chem 2007;14:1956–1965.
Ravindranath MH, Muthugounder S, Saravanan TS, et al. Human antiganglioside autoantibodies: validation of ELISA. Ann NY Acad Sci 2005;1050:229–242.
Miller CC, Ackerley S, Brownlees J, et al. Axonal transport of neurofilaments in normal and disease states. Cell Mol Life Sci 2002;59:323–330.
Rao MV, Nixon RA . Defective neurofilament transport in mouse models of amyotrophic lateral sclerosis: a review. Neurochem Res 2003;28:1041–1047.
Forooghian F, Cheung RK, Smith WC, et al. Enolase and arrestin are novel nonmyelin autoantigens in multiple sclerosis. J Clin Immunol 2007;27:388–396.
Demakova EV, Korobov VP, Lemkina LM . Determination of gamma-aminobutyric acid concentration and activity of glutamate decarboxylase in blood serum of patients with multiple sclerosis. Klin Lab Diagn 2003;4:15–17.
Raska M, Weigl E . Heat shock proteins in autoimmune diseases. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2005;149:243–249.
Matute C, Domercq M, Sanchez-Gomez MV . Glutamate-mediated glial injury: mechanisms and clinical importance. Glia 2006;53:212–224.
Lipton SA, Rosenberg PA . Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 1994;330:613–622.
Pitt D, Nagelmeier IE, Wilson HC, et al. Glutamate uptake by oligodendrocytes: implications for excitotoxicity in multiple sclerosis. Neurology 2003;61:1113–1120.
Vercellino M, Merola A, Piacentino C, et al. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 2007;66:732–739.
Srinivasan R, Sailasuta N, Hurd R, et al. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain 2005;128:1016–1025.
Stover JF, Pleines UE, Morganti-Kossmann MC, et al. Neurotransmitters in cerebrospinal fluid reflect pathological activity. Eur J Clin Invest 1997;27:1038–1043.
Sarchielli P, Greco L, Floridi A, et al. Excitatory amino acids and multiple sclerosis: evidence from cerebrospinal fluid. Arch Neurol 2003;60:1082–1088.
Choi DW . Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett 1985;58:293–297.
Newcombe J, Uddin A, Dove R, et al. Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol 2008;18:52–61.
Stys PK, Lipton SA . White matter NMDA receptors: an unexpected new therapeutic target? Trends Pharmacol Sci 2007;28:561–566.
Wallstrom E, Diener P, Ljungdahl A, et al. Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci 1996;137:89–96.
Bolton C, Paul C . MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J Pharmacol Exp Ther 1997;282:397–402.
Smith T, Groom A, Zhu B, et al. Autoimmune encephalomyelitis ameliorated byAMPA antagonists. Nat Med 2000;6:62–66.
Groom AJ, Smith T, Turski L . Multiple sclerosis and glutamate. Ann NY Acad Sci 2003;993:229–275.
Whitney KD, McNamara JO . GluR3 autoantibodies destroy neural cells in a complement-dependent manner modulated by complement regulatory proteins. J Neurosci 2000;20:7307–7316.
Xiong ZQ, Qian W, Suzuki K, et al. Formation of complement membrane attack complex in mammalian cerebral cortex evokes seizures and neurodegeneration. J Neurosci 2003;23:955–960.
Mantegazza R, Bernasconi P, Baggi F, et al. Antibodies against GluR3 peptides are not specific for Rasmussen's encephalitis but are also present in epilepsy patients with severe, early onset disease and intractable seizures. J Neuroimmunol 2002;131:179–185.
Levite M . Autoimmune epilepsy. Nat Immunol 2003;3:500.
Watson R, Jiang Y, Bermudez I, et al. Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology 2004;63:43–50.
Dropcho EJ . Principles of paraneoplastic syndromes. Ann NY Acad Sci 1998;841:246–261.
Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25–36.
Iizuka T, Sakai F, Ide T, et al. Anti-NMDA receptor encephalitis in Japan: long-term outcome without tumor removal. Neurology 2008;70:504–511.
Storstein A, Knudsen A, Vedeler CA . Proteasome antibodies in paraneoplastic cerebellar degeneration. J Neuroimmunol 2005;165:172–178.
Feist E, Dörner T, Kuckelkorn U, et al. Diagnostic importance of anti-proteasome antibodies. Int Arch Allergy Immunol 2000;123:92–97.
Wright CF, Oswald BW, Dellis S . Vaccinia virus late transcription is activated in vitro by cellular heterogeneous nuclear ribonucleoproteins. J Biol Chem 2001;276:40680–40686.
Ducray F, Roos-Weil R, Garcia PY, et al. Devic's syndrome-like phenotype associated with thymoma and anti-CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry 2007;78:325–327.
Camdessanché JP, Lassablière F, Meyronnet D, et al. Expression of the onconeural CV2/CRMP5 antigen in thymus and thymoma. J Neuroimmunol 2006;174:168–173.
Cross SA, Salomao DR, Parisi JE, et al. Paraneoplastic autoimmune optic neuritis with retinitis defined by CRMP-5-IgG. Ann Neurol 2003;54:38–50.
Lees F, MacDonald A-M, Turner JWA . Leber's disease with symptoms resembling disseminated sclerosis. J Neurol Neurosurg Psychiatry 1964;27:415–421.
Harding AE, Riordan-Eva P, Govan GG . Mitochondrial DNA diseases: genotype and phenotype in Leber's hereditary optic neuropathy. Muscle Nerve 1995;3:S82–S84.
Smith PR, Cooper JM, Govan GG, et al. Antibodies to human optic nerve in Leber's hereditary optic neuropathy. J Neurol Sci 1995;130:134–138.
Ciric B, Van Keulen V, Paz Soldan M, et al. Antibody-mediated remyelination operates through mechanism independent of immunomodulation. J Neuroimmunol 2004;146:153–161.
Acknowledgements
BK is supported by the National Multiple Sclerosis Society (RG3521A6/T, PP1334), Serono and Veterans Administration.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vyshkina, T., Kalman, B. Autoantibodies and neurodegeneration in multiple sclerosis. Lab Invest 88, 796–807 (2008). https://doi.org/10.1038/labinvest.2008.53
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2008.53
Keywords
This article is cited by
-
The Role of Glutamate in the Pathogenesis of Multiple Sclerosis
Neuroscience and Behavioral Physiology (2020)
-
Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics
BMC Medicine (2013)
-
Avidity of anti-neurocytoskeletal antibodies in cerebrospinal fluid and serum
Folia Microbiologica (2012)
