
Abstract
Mitochondrial toxicity results from pyrimidine nucleoside reverse transcriptase inhibitors (NRTIs) for HIV/AIDS. In the heart, this can deplete mitochondrial (mt) DNA and cause cardiac dysfunction (eg, left ventricle hypertrophy, LVH). Four unique transgenic, cardiac-targeted overexpressors (TGs) were generated to determine their individual impact on native mitochondrial biogenesis and effects of NRTI administration on development of mitochondrial toxicity. TGs included cardiac-specific overexpression of native thymidine kinase 2 (TK2), two pathogenic TK2 mutants (H121N and I212N), and a mutant of mtDNA polymerase, pol-γ (Y955C). Each was treated with antiretrovirals (AZT-HAART, 3 or 10 weeks, zidovudine (AZT) + lamivudine (3TC) + indinavir, or vehicle control). Parameters included left ventricle (LV) performance (echocardiography), LV mtDNA abundance (real-time PCR), and mitochondrial fine structure (electron microscopy, EM) as a function of duration of treatment and presence of TG. mtDNA abundance significantly decreased in Y955C TG, increased in TK2 native and I212N TGs, and was unchanged in H121N TGs at 10 weeks regardless of treatment. Y955C and I212N TGs exhibited LVH during growth irrespective of treatment. Y955C TGs exhibited cardiomyopathy (CM) at 3 and 10 weeks irrespective of treatment, whereas H121N and I212N TGs exhibited CM only after 10 weeks AZT-HAART. EM features were consistent with cardiac dysfunction. mtDNA abundance and cardiac functional changes were related to TG expression of mitochondrially related genes, mutations thereof, and NRTIs.
Similar content being viewed by others


Main
Mitochondrial biogenesis is defined as the control of mitochondrial turnover, content, and number to maintain integrity and tissue energy demands.1 In the heart, mitochondria are abundant; they constitute approximately 40% of total cardiomyocyte volume and produce more than 90% of its energy.2 Because mitochondria possess their own unique DNA (mtDNA; reviewed in Pakendorf and Stoneking3), altered mtDNA replication can result in diminished mitochondrial biogenesis, decreased energy output, and organ dysfunction.4 Side effects of antiretroviral drugs include defective mitochondrial biogenesis5 and a relationship to inhibition of pol-γ by nucleoside reverse transcriptase inhibitors (NRTIs) has been posited.6 Pyrimidine NRTIs used in HIV/AIDS compete with native nucleotides for incorporation into nascent mtDNA. It was hypothesized that temporal changes may be associated with growth and physiological end points as well as molecular thresholds of mtDNA toxicity that could be related to specific gene-targeted mutations.
To define mtDNA replication defects in vivo, genes encoding polypeptides integral to mitochondrial biogenesis were transgenically targeted (TG) to the murine heart. Treatment with NRTIs parametrically defined effects on cardiac mtDNA replication and function in TGs and wild-type (WT) littermates.
Cardiac-targeted TGs for thymidine kinase 2 (TK2) were used. TK2 is a mitochondrial pyrimidine deoxynucleoside salvage enzyme involved in pyrimidine nucleoside phosphorylation in mitotically quiescent cells.7 Overexpression of native TK2, and two mutants of TK2 (H121N and I212N; known to cause ‘inherited mtDNA depletion syndromes’8) were used. The TK2 mutant TGs served as ‘dominant negatives’ that interrupt pyrimidine nucleoside phosphorylation homeostasis.
Another cardiac-targeted TG expressed a mutant (Y955C) of the DNA polymerase-γ (pol-γ) uniquely responsible for mtDNA replication. Y955C is the commonest, most severe autosomal dominant POLG mutation.9, 10 This TG exhibited severely decreased mtDNA, increased oxidative stress, and cardiomyopathy (CM),11 whereas more recent studies demonstrated mtDNA depletion and CM after 5 week AZT-HAART.12
Owing to the duration of these studies, it was important to establish changes (if any) that related to murine growth. Temporal changes in mtDNA abundance during early adulthood were defined in FVB/n WT mice. mtDNA abundance and cardiac dysfunction were subsequently determined in cardiac-targeted TGs expressing above-mentioned genes and AZT-HAART pharmacological treatment. These experiments tested whether genetically regulated differences in mtDNA replication along with pharmacological manipulation impact cardiac mtDNA replication and performance.
MATERIALS AND METHODS
α-MyHc Promoter-Driven Mutant TGs
Cardiac-targeted overexpressing transgenic mouse lines were used. Multiple lines for each TG were previously generated for native TK2 TG,13 TK2 mutant (H121N and I212N) TGs12, 14 and Y955C pol-γ mutant TG.11 Relative copy number for each line was determined. Low copy number lines were selected for these studies to prevent nonspecific strong overexpression effects.15 No changes in cognitive behavior, growth, maturation, breeding behavior, or Mendelian distribution were found in any of the lines. TG mice were routinely identified for each generation using dot blot analysis and real-time PCR.11
Treatment Protocols
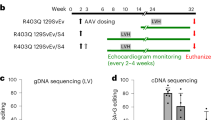
Procedures complied with Emory IACUC and NIH guidelines. WT and TG littermates (male and female) were age-matched (8–12 weeks) at the start of the treatment regimen. Cohorts of 15–20 mice were used in standard ‘2 × 2’ factorial protocols. Food and water were provided ad libitum. Antiretroviral drugs were from the manufacturers (compliments of Professor Raymond Schinazi, VA Medical Center, Decatur, GA and Emory Center for AIDS Research Pharmacology Core). Dosing was done by daily gavage (morning) at doses that resemble human therapy on an mg/kg per day basis. Combination antiretroviral treatments included zidovudine (AZT; 0.22 mg per day) together with lamivudine (3TC; 0.11 mg per day) and indinavir (IDV; 0.9 mg per day) or vehicle controls. Morbidity and mortality from the procedure were negligible. After 3 and 10 weeks of treatment (11–13 or 19–22 weeks of age), body weights and echocardiographic (ECHO) measurements were carried out, animals were terminated, and samples were retrieved and stored at −80°C for DNA extraction and analysis (Figure 1).

Timeline of longitudinal studies. Treatment course is outlined with equivalent biological age indicated and overall range of maturation phase vs adulthood shown (based on Figure 2 data).
mtDNA and Nuclear DNA Quantitation in Heart and Skeletal Muscle Tissue Using Real-Time PCR
Methods used were modifications of those from others16 as employed by us in the past.11, 13 Total DNA was extracted from heart and skeletal muscle tissue (∼10 mg wet weight) using a MagNA Pure System and reagents (Roche Life Sciences, Indianapolis, IN, USA). DNA sequences for primers and probes used for quantitation of mitochondrial and nuclear DNA (nDNA) were described.13 Real-Time PCR was performed in duplicate for each amplicon. Amplification was performed using LC 480 (Roche Life Sciences). Standard DNA curves for quantitation of the LC products were used. PCR products of mtDNA and nDNA were quantified using the corresponding external standard.
Echocardiography in Mutant TGs and WT
ECHO was performed at experimental termination. Left ventricular (LV) mass and LV end-diastolic dimension (LVEDD) were quantified and normalized using ECHO in age- and gender-matched (littermate) WT and TGs as before.17 Mice were anesthetized with Avertin (0.25 mg/g of body weight) and two-dimensionally targeted M-mode images were generally taken from the short axis (at the level of the largest LV diameter) by using a 15 MHz transducer (Acuson SequoiaTM; Siemens Medical Solutions USA Inc., Malvern, PA, USA).
Fine structure pathological evaluations with transmission electron microscopy analysis of mitochondrial damage of mutant TGs
Ultrastructure (N=12) was evaluated with electron microscopy (EM) using methods used regularly in the laboratory.18 Sections (0.5 μm) were cut with glass knives and stained with toluidine blue for orientation. Ultrathin (900 Å) sections were cut with a diamond knife, stained with uranyl acetate and lead citrate, and examined by EM (Philips Morgagni Model 201; Philips, Eindhoven, Amsterdam). They were also evaluated and photographed. Each EM photomicrograph was reviewed independently by two investigators. Parameters included presence of structurally abnormal mitochondria, increased numbers of mitochondrial profiles per field, intramitochondrial lamellar bodies, abnormal cristae density, cristae reduplication, mitochondrial swelling, and intramitochondrial paracrystals.5
mtDNA Density
Changes in mtDNA were normalized to LV mass. The ratio was used to define an index of mitochondrial density in the heart. Ratios of cardiac mtDNA abundance to LV mass were calculated (mtDNA density) for each individual mouse. Mean mtDNA densities were determined for each cohort and changes from WTs were calculated for each TG and treatment. Temporal changes in mtDNA density were also determined for each WT and TG (3 weeks compared to 10 weeks).
Statistical Analysis
Data were expressed as the ratio of mean values for mtDNA to the mean values of nDNA × 10−3. Resultant values were expressed as mean±standard error, normalized to untreated WT mean (set at 1.0). A value of P<0.05 was considered statistically significant. Echocardiographic determinations from all groups were compared by one-way ANOVA.17
RESULTS
Age-Related Changes in mtDNA
Wild-type FVB/n mice were assessed for mtDNA abundance in skeletal and cardiac muscle tissues at intervals from 7 to 26 weeks (a critical period for development,19 somewhat analogous in humans to young adulthood). Results defined changes in mtDNA abundance associated with this time of continued growth and increase in muscle mass between 7 and 16 weeks of age (Figure 1). Skeletal muscle demonstrated a step-wise increase in mtDNA/nDNA ratio from 7 to 13 weeks, and again at 16–18 weeks (Figure 2a). Cardiac muscle increased in mtDNA abundance between 13 weeks and 16–18 weeks (Figure 2b). mtDNA/nDNA reached a plateau (2.8±0.2) at 16–18 weeks and stabilized to the 26-week end point.

Temporal changes in mtDNA abundance during maturation to adult. Skeletal muscle and cardiac tissues (a and b, respectively) were collected from WT FVB/n cohorts at various end points in early age beginning at weeks 7–9 (youth) at intervals up to 26 weeks (adult).
Temporal Changes in mtDNA Abundance Following Treatment with AZT-HAART
On the basis of above, 8- 10-week-old mice were used to define NRTI effects. Mice were treated with AZT-HAART or vehicle for 3 or 10 weeks. This corresponds to cardiac growth (11–13 weeks) or mature adult murine heart (19–22 weeks), at termination (Figure 1). mtDNA abundance was determined in cardiac tissues of WT mice and each of five different TGs with and without antiretrovirals.
TK2 native and mutant TGs demonstrated three distinct outcomes. At 3 weeks, vehicle-treated TK2 native TGs exhibited control levels of mtDNA whereas TG littermates combined with AZT-HAART resulted in increased cardiac mtDNA abundance (Figure 3a). Alternatively, mutant TK2 TGs (H121N and I212N) alone also exhibited increased mtDNA in myocardial extracts at 3 weeks, in this case independent of AZT-HAART treatment. After 10 weeks, TK2 native TGs increased mtDNA abundance independent of AZT-HAART treatment, suggesting a transgenic effect (Figure 3b). I212N TK2 TGs continued to demonstrate increased mtDNA abundance as a transgenic effect, independent of AZT-HAART (Figure 3b). In contrast, H121N TGs exhibited control levels of mtDNA in both vehicle- and AZT-HAART-treated cohorts compared to WT littermates at 10 weeks (Figure 3b).

Longitudinal changes in relative mtDNA abundance after AZT-HAART treatment initiation. Using ‘2 × 2’ protocols, four different TGs were treated with AZT-HAART or vehicle for 3 and 10 weeks duration (a and b, respectively) and mtDNA abundance levels from cardiac tissues were determined and compared to WT littermates.
Unlike the increase in mtDNA that resulted in TK2 native and mutant TGs, Y955C TGs consistently demonstrated significantly decreased mtDNA abundance in myocardial extracts (<50%, P<0.05) compared to WTs at both 3 and 10 weeks (Figure 3a and b). This decrease in mtDNA occurred independent of AZT-HAART, suggesting the mutant transgene alone was responsible for the marked decrease in mtDNA abundance. The absolute decrease in mtDNA abundance induced in the Y955C TGs (vehicle treated) worsened over time (0.5 at 3 weeks to 0.3 at 10 weeks; ∼19% change).
Cardiac Function in TG Cohorts
Cardiac function was assessed echocardiographically for each group of TG and WT mice at 3 and 10 weeks as a function of AZT-HAART treatment. Both LV mass and LVEDD were determined. The impact of each TG on cardiac function at 3 weeks varied step-wise from no change to prominent left ventricle hypertrophy (LVH), depending on the nature of the specific TG. TK2 native overexpressor TG LV mass was comparable to WT (Figure 4a). H121N mutant TK2 had no effect. When AZT-HAART was added, TGs exhibited a small but significant LV mass increase (Figure 4a). I212N mutant TK2 TGs exhibited LVH, independent of AZT-HAART (Figure 4a). Likewise, Y955C TGs exhibited LVH with the greatest change in LV mass among all the TG cohorts. In addition, Y955C LVH was further exacerbated by AZT-HAART (Figure 4a), suggesting a synergy of effects induced by both the transgene and NRTI treatment.

Longitudinal cardiac hypertrophy development in TGs. Left ventricular mass (LV mass) was recorded for ‘2 × 2’ cohorts of TGs treated with AZT-HAART or vehicle at 3 and 10 weeks duration (a and b, respectively).
An interesting outcome occurred after 10 weeks. Independent of the specific transgene, all TG cohorts demonstrated LVH that was further amplified by AZT-HAART (Figure 4b). Even WT cohorts treated with AZT-HAART exhibited LVH compared to vehicle-treated littermates.
LVEDD remained unchanged in TG cohorts at 3 weeks. The exception was Y955C (Figure 5a) where LV dysfunction and LV dilation occurred early and persisted to 10 weeks. LVEDD increased in H121N and I212N TGs when combined with AZT-HAART (Figure 5b). Although LVH occurred in all TGs with or without AZT-HAART at 10 weeks, dilation occurred only in TK2 mutant TGs (ie, native TK2) after 10 weeks of AZT-HAART.

Longitudinal development of cardiac dilation in TGs. Left ventricular end-diastolic dimension (LVEDD) was determined for TGs and WTs treated with AZT-HAART or vehicle at 3 and 10 weeks duration (a and b, respectively).
EM Features of Mitochondria in TG Hearts
EM changes in cardiac mitochondria of the TGs were defined. Because LVH was found in all TGs and WTs treated with AZT-HAART at 10 weeks, observable ultrastructural changes were expected. Vehicle-treated WTs demonstrated normal, characteristic, oval mitochondria that contained densely packed cristae (Figure 6a). AZT-HAART-treated WTs maintained normal mitochondrial size and shape but with less prominent and discernable cristae (Figure 6b), indicating an NRTI-treatment effect on WT mitochondrial structure after 10 weeks. Vehicle-treated TK2 TGs (native TK2, and H121N or I212N mutants) exhibited mitochondria of normal size with essentially no detectable changes when compared to WTs (Figure 6a). H121N and I212N TK2 mutant TGs had relatively few changes following AZT-HAART treatment, whereas native TK2 TGs demonstrated irregular shaped mitochondria with less prominent, truncated cristae that resembled that of WTs undergoing similar treatment (Figure 6b). Vehicle-treated Y955C TGs exhibited increased mitochondrial number, irregular shape, and sparse fragmented cristae (Figure 6a). Additive ultrastructural damage to Y955C TGs following AZT-HAART was not easily discernable. This could result from the ‘high background’ damage in the vehicle-treated TGs (Figure 6b).

Ultrastructural changes in cardiac mitochondrial tissues following 10 weeks of AZT-HAART. Cardiac tissues from all four TG cohorts treated with vehicle (a) or AZT-HAART (b) and WT littermates were compared using electron microscopy (EM, original magnification × 22 400).
Changes in mtDNA Density
Cumulative mtDNA abundance and cardiac functional data were complex. To summarize the data cohesively and integrate these parameters, a mathematical relationship between mtDNA abundance and LV mass was calculated. This derived number was operationally defined as ‘mtDNA density’. Resultant calculations (Table 1) clarified functional and molecular trends and phenotypic changes. Data from 3 weeks of AZT-HAART revealed no significant change in mtDNA density for native TK2 overexpressors when compared to those of WTs. However, both vehicle- and all AZT-HAART-treated mutant TGs (Y955C, H121N, and I212N) exhibited significant changes. Vehicle- and AZT-HAART-treated Y955C TGs each exhibited decreased mtDNA density (63–79%). In contrast, H121N and I212N mtDNA densities increased (60–90%) over WTs.
Following 10 weeks AZT-HAART, TK2 TGs demonstrated increased mtDNA density (33–74%). This resembled findings in I212N TGs in which increased mtDNA density at 10 weeks persisted (P<0.001). Y955C TG cardiac mtDNA density decreased further (P<0.001) at 10 weeks. A variation occurred in the case of H121N TGs. At 10 weeks, mtDNA density for vehicle-treated H121N TGs resembled that of WTs, however, AZT-HAART-treated H121N TGs exhibited decreased mtDNA density (46%) compared to AZT-HAART-treated WT controls. Temporal changes (3 vs 10 weeks) were small overall. Exceptions included the vehicle-treated native TK2 TGs in which mtDNA density increased at 10 weeks compared to 3 weeks (P<0.05) and H121N TGs in which mtDNA density decreased at 10 weeks compared to 3 weeks (P<0.001).
DISCUSSION
Normal cardiac function depends on adequate mitochondrial energy production.20 TGs were used to interrupt nucleoside homeostasis and mtDNA replication selectively. Results revealed that animal maturation and molecular factors impact mitochondrial biogenesis and cardiac function.
Biological effects of age (in maturation to adulthood) were explored in parallel to pharmacological treatment. A ‘growth-related’ increase in mtDNA abundance was found in both skeletal and cardiac muscle tissues of the parental FVB/N strain (7–9 weeks up to 16–18 weeks of age). Increased mtDNA abundance correlated with muscle maturation. Different murine strains may respond to cardiac stimuli in different ways.21 TK2 and pol-γ is well conserved between rodentic strains so the likelihood of strain-specific effects at that level is small. Nonetheless, data from these experiments may vary somewhat to data obtained on other inbred murine strains. Nevertheless, the biological relevance of growth-related changes in mtDNA abundance remains unclear and is worthy of further investigation.
Regulation of mitochondrial pathways allows organisms to meet short-term changes in energy demand. When metabolic demand persists, remodeling of mitochondrial structure and function occurs to accommodate such needs (reviewed in Moyes and Hood22). This process is integral to developmental, physiological, and pathological conditions including myogenesis, development, exercise, and environmental stress.
Of the TGs examined here, Y955C caused the worst defects in cardiac mtDNA replication both in amplitude and rapidity of onset. In parallel, cardiac dysfunction was substantial, and was worsened by NRTIs. Interestingly, GFP overexpression driven by α-myosin heavy chain promoter suggested that transgenic overexpression alone could induce dilated CM.23 A factor in that study was the ‘dose-dependent effect’. Only two of the TG lines with highest expression exhibited CM. Conversely, low copy number TG lines used here demonstrated survival comparable to WTs.11, 12, 13 In addition, native TK2 TGs exhibited neither CM nor changes in mtDNA at 3 weeks compared to Y955C TGs. It may be possible to consider cardiac-targeted native pol-γ TG could serve as an important control, but this TG was not available at the time of these studies. Data here support the concept that pathogenetic mutations in pol-γ profoundly impact mitochondrial homeostasis and yield a CM phenotype.24
TK2 (located intramitochondrially) is the primary kinase of pyrimidine nucleosides. TK2 has no catalytic activity on purines.25 Cardiac-targeted TK2 TG mutants exhibit reduced activity.8 The potential impact of TK2 (either native or mutant) results in cardiac dysfunction by 10 weeks of AZT-HAART. With native TK2, it is plausible that increased TK2 mass could disrupt the intramitochondrial substrate (nucleotide) pool balance or availability of pol-γ nucleotide substrate and lead to changes in mtDNA abundance at the level of substrate. DNA precursor pool asymmetries in mammalian mitochondria have been proposed to contribute to mtDNA mutagenesis through reduced mtDNA replication fidelity.26, 27 Nucleotide pool changes brought on by overexpression of TK2 could lead to altered mtDNA replication fidelity.
The disparate outcomes on mtDNA abundance associated with TK2-targeted TGs (increased mtDNA abundance) in contrast to pol-γ TG (decreased mtDNA abundance) reinforce clinical observations and disparate outcomes that occur in human patients undergoing identical treatments. The underlying implication is that genetic factors such as unique mutations in key genes involved in mtDNA replication can impact the overall mtDNA biogenesis in different ways. Depending on the specific mutation, changes in pathophysiological phenotype (ie, cardiac function) may exhibit early onset (ie, 3 weeks in FVB/n), be delayed until a threshold is reached (ie, 10 weeks in FVB/n), or remain undetected. In some cases, an existing genetic mutation may have no impact alone, but when combined with a pharmacological agent (eg, NRTI) reach the critical threshold through a synergistic effect that renders a phenotypic change. The ‘gene–environment’ interaction has implications for ‘personalized medicine’.28, 29
In summary, targeting proteins involved in mtDNA replication yielded distinct effects on cardiac mtDNA abundance and myocardial function (Figure 7). Y955C disrupted mtDNA replication early. Neither mutations in TK2 resulted in early mtDNA changes, but long-term changes resulted. Mutant TGs (Y955C, H121N, and I212N) had greater impact on changes in mtDNA abundance and cardiac dysfunction than native TK2.

Schematic summary of the impact of diverse mutant TGs of mtDNA replication pathway. Highlighted are the roles of native TK2 or TK2 mutants in the phosphorylation of nucleoside analogues (eg, AZT) and the role of pol-γ mutant (Y955C) in the incorporation of the active nucleoside triphosphate (eg, AZT-TP) in mtDNA replication. These constitute potential targets for inhibition of the mtDNA replication pathway.
References
McLeod CJ, Pagel I, Sack MN . The mitochondrial biogenesis regulatory program in cardiac adaptation to ischemia—a putative target for therapeutic intervention. Trends Cardiovasc Med 2005;15:118–123.
Barth E, Stammler G, Speiser B, et al. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 1992;24:669–681.
Pakendorf B, Stoneking M . Mitochondrial DNA and human evolution. Annu Rev Genomics Hum Genet 2005;6:165–183.
Wallace DC . Diseases of the mitochondrial DNA. Annu Rev Biochem 1992;61:1175–1212.
Dalakas MC, Illa I, Pezeshkpour GH, et al. Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med 1990;322:1098–1105.
Lewis W, Dalakas MC . Mitochondrial toxicity of antiviral drugs. Nat Med 1995;1:417–422.
Eriksson S, Munch-Petersen B, Johansson K, et al. Structure and function of cellular deoxyribonucleoside kinases. Cell Mol Life Sci 2002;59:1327–1346.
Saada A, Shaag A, Mandel H, et al. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 2001;29:342–344.
Copeland WC . Inherited mitochondrial diseases of DNA replication. Annu Rev Med 2008;59:131–146.
Graziewicz MA, Longley MJ, Copeland WC . DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev 2006;106:383–405.
Lewis W, Day BJ, Kohler JJ, et al. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab Invest 2007;87:326–335.
Kohler JJ, Hosseini SH, Green E, et al. Cardiac-targeted transgenic mutant mitochondrial enzymes: mtDNA defects, antiretroviral toxicity and cardiomyopathy. Cardiovasc Toxicol 2008;8:57–69.
Hosseini SH, Kohler JJ, Haase CP, et al. Targeted transgenic overexpression of mitochondrial thymidine kinase (TK2) alters mitochondrial DNA (mtDNA) and mitochondrial polypeptide abundance: transgenic TK2, mtDNA, and antiretrovirals. Am J Pathol 2007;170:865–874.
Wang J, Su C, Neuhard J, et al. Expression of human mitochondrial thymidine kinase in Escherichia coli: correlation between the enzymatic activity of pyrimidine nucleoside analogues and their inhibitory effect on bacterial growth. Biochem Pharmacol 2000;59:1583–1588.
Lefai E, Calleja M, Ruiz de Mena I, et al. Overexpression of the catalytic subunit of DNA polymerase gamma results in depletion of mitochondrial DNA in Drosophila melanogaster. Mol Gen Genet 2000;264:37–46.
Cote HC, Yip B, Asselin JJ, et al. Mitochondrial:nuclear DNA ratios in peripheral blood cells from human immunodeficiency virus (HIV)-infected patients who received selected HIV antiretroviral drug regimens. J Infect Dis 2003;187:1972–1976.
Lewis W, Haase CP, Raidel SM, et al. Combined antiretroviral therapy causes cardiomyopathy and elevates plasma lactate in transgenic AIDS mice. Lab Invest 2001;81:1527–1536.
Lewis W, Grupp IL, Grupp G, et al. Cardiac dysfunction occurs in the HIV-1 transgenic mouse treated with zidovudine. Lab Invest 2000;80:187–197.
Thomopoulos S, Kim HM, Rothermich SY, et al. Decreased muscle loading delays maturation of the tendon enthesis during postnatal development. J Orthop Res 2007;25:1154–1163.
Katz AM . Is the failing heart energy depleted? Cardiol Clin 1998;16:633–644, viii.
Hoit BD, Kiatchoosakun S, Restivo J, et al. Naturally occurring variation in cardiovascular traits among inbred mouse strains. Genomics 2002;79:679–685.
Moyes CD, Hood DA . Origins and consequences of mitochondrial variation in vertebrate muscle. Annu Rev Physiol 2003;65:177–201.
Huang WY, Aramburu J, Douglas PS, et al. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat Med 2000;6:482–483.
Copeland WC . Inherited mitochondrial diseases of DNA replication. Annu Rev Med 2008;59:131–146.
Eriksson S, Munch-Petersen B, Kierdaszuk B, et al. Expression and substrate specificities of human thymidine kinase 1, thymidine kinase 2 and deoxycytidine kinase. Adv Exp Med Biol 1991;309B:239–243.
Song S, Pursell ZF, Copeland WC, et al. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc Natl Acad Sci USA 2005;102:4990–4995.
Ferraro P, Nicolosi L, Bernardi P, et al. Mitochondrial deoxynucleotide pool sizes in mouse liver and evidence for a transport mechanism for thymidine monophosphate. Proc Natl Acad Sci USA 2006;103:18586–18591.
Davies SM . Pharmacogenetics, pharmacogenomics and personalized medicine: are we there yet? Hematology Am Soc Hematol Educ Program 2006;1:111–117.
Nebert DW, Zhang G, Vesell ES . From human genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug Metab Rev 2008;40:187–224.
Acknowledgements
This work was supported by NIH NHLBI R01 HL059798 and HL079867 (to WL) and NIDDK K01 DK078513 (to JK).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kohler, J., Hosseini, S., Cucoranu, I. et al. Murine cardiac mtDNA: effects of transgenic manipulation of nucleoside phosphorylation. Lab Invest 89, 122–130 (2009). https://doi.org/10.1038/labinvest.2008.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2008.121
Keywords
This article is cited by
-
Aging and HIV/AIDS: pathogenetic role of therapeutic side effects
Laboratory Investigation (2014)
-
Transgenic mouse model with deficient mitochondrial polymerase exhibits reduced state IV respiration and enhanced cardiac fibrosis
Laboratory Investigation (2013)
-
Tenofovir renal proximal tubular toxicity is regulated By OAT1 and MRP4 transporters
Laboratory Investigation (2011)
-
Transgenic cardiac-targeted overexpression of human thymidylate kinase
Laboratory Investigation (2010)
-
Absence of Mitochondrial Toxicity in Hearts of Transgenic Mice Treated with Abacavir
Cardiovascular Toxicology (2010)
