
Abstract
Basal cell nevus syndrome (BCNS or Gorlin syndrome, OMIM: 109400) is a rare autosomal dominant disorder with high penetrance. It is characterized by developmental anomalies and predisposition to tumors (for example, basal cell carcinoma (BCC) and medulloblastoma). PTCH1, the human homolog of the Drosophila patched gene, was identified as a gene responsible for BCNS. The PTCH1 protein is a Hedgehog (Hh) protein receptor and is pivotal for early development, stem cell maintenance and/or differentiation. We analyzed the six Japanese families with BCNS and identified six germline mutations in the PTCH1 gene. One family had a nonsense mutation (c.1196G>A), one had a 1-bp deletion (c.2029delA), two had 2-bp deletions (c.239_240delGA and c.1670_1671delCA) and one had a 58-bp duplication (c.1138_1195dup). They caused premature termination, resulting in the truncation of the PTCH1 protein. Analysis of a high-density single nucleotide polymorphism (SNP) mapping array showed a large ∼1.2-Mb deletion, including the PTCH1 gene in one allele, in a family in which PTCH1 mutations were not identified at the sequence level. These data indicated that all the six families who were diagnosed with BCNS had mutations in the PTCH1 gene and that a single copy of a PTCH1 mutation causes BCNS.
Similar content being viewed by others

Introduction
Basal cell nevus syndrome (BCNS, OMIM: 109400), also known as Gorlin syndrome or Nevoid basal cell carcinoma syndrome, is a rare autosomal dominant disorder with high penetrance.1 BCNS is characterized by developmental abnormalities, such as jaw keratocyst, calcification of the falx cerebri, multiple nevi, palmar and plantar pits and skeletal anomalies.2, 3 It is also known to predispose individuals to cancers, including basal cell carcinoma (BCC), medulloblastoma, rhabdomyosarcoma and benign tumors, such as ovarian fibroma and cardiac fibroma. The Hedgehog (Hh) receptor Patched1 (PTCH1) gene was identified as a gene responsible for BCNS.4, 5 Hh signaling has important roles during early development as well as during postembryonic development,6, 7 and disruption of the signaling cascade induces several diseases in humans.8, 9 Repression of Hh signaling causes malformations, such as holoprosencephaly and polydactyly, and aberrant activation of the cascade induces proliferation and malignant transformation, including medulloblastoma, BCC and various digestive organ tumors.10 Hh signaling activation due to impaired PTCH1 function was reported in BCNS and also among malignant neoplasms, such as sporadic BCC, medulloblastoma/primitive neuroectodermal tumor, breast cancer, colon cancer and meningioma.11
Human PTCH1, a homolog of the Drosophila segment polarity gene patched (ptc), has at least 23 exons spanning ∼50 kb and is located on chromosome 9q22.32. It encodes an integral membrane protein of 1447 amino acids.4, 5 The PTCH1 protein has 12 potential hydrophobic membrane-spanning domains, intracellular amino- and carboxy-terminal regions and two large hydrophilic extracellular loops in which Hh proteins bind.12, 13
Identification of mutations in PTCH1 is very important for the clinical management of the BCNS patient's family, because patients with mutations have a high risk of BCC and other cancers for the rest of their lives. Furthermore, radiation treatment for benign tumors in patients with BCNS induces more aggressive tumors.14 Thus, for preventing unnecessary development of tumors, it is essential to diagnose BCNS in early childhood. To date, ∼300 mutations, including nonsense and missense mutations, insertions, deletions and splicing mutations, have been identified in the PTCH1 gene.11 Half of the mutations were reported in BCNS cases and the rest were in sporadic tumors, including BCC and medulloblastoma. However, the majority of the data were obtained from Caucasian BCNS patients. Although no strong correlation between the mutated region and patients’ phenotypes has been shown,15 it is valuable to analyze correlations between the types of mutation in the PTCH1 gene, the genetic or epigenetic background of the patients and the clinical features of BCNS. In this study, we report novel small mutations and one large deletion of the PTCH1 gene in six Japanese BCNS families.
Materials and methods
Subjects
The subjects were 15 Japanese individuals from six unrelated families who visited the Osaka Dental University and Hyogo College of Medicine, Japan, in the early 1980s and in 2008 (Figure 1). Five children of 5–10 years of age (NS1, 2, 3, 6 and 9) (NS refers to the test subjects), and an adult in her late twenties (NS5), were diagnosed with BCNS on the basis of the presence of multiple jaw keratocytes and characteristic facial appearances, such as macrocephaly and coarse facial features, with or without calcification of the falx. The diagnostic criterion2 in current use was not applied, as it was not available at that time. Three further BCNS cases (NS4, 7 and 8) were identified during searches of the probands’ families. None of the six probands, their siblings or their parents had developmental disorders or skin cancers. A sibling of NS9 died of a brain tumor at the age of 2 years. The parents and adult patients gave informed consent to undergo cytogenetic analysis and radiation sensitivity tests using blood cells and fibroblasts. None of them showed hypersensitivity to ultraviolet (UV) radiation on the basis of the analysis of surviving cells after UV irradiation. All cells, except NS9-derived cells, were stored anonymously. The NS9 patient visited our hospital for genetic counseling 20 years after the initial diagnosis of BCNS and gave written informed consent for participation in this study. The child of this patient (NS10) was also examined under informed consent of the parents. This work was approved by the Medical Ethical Committee of Hyogo College of Medicine.

Pedigrees of six families with basal cell nevus syndrome (BCNS). Filled symbols indicate patients. Asterisks (*) indicate unaffected individuals who underwent molecular analysis. The arrows represent the probands.
DNA isolation, amplification and sequencing analysis
Genomic DNAs from Epstein–Barr virus-immortalized lymphoblastoid cells, skin fibroblasts or blood were extracted using a QIAamp DNA Blood Micro Kit (Qiagen, Hilden, Germany). All coding exons and intron–exon boundaries were PCR amplified and sequenced. The majority of the primers were designed according to Fujii et al.16 and are summarized in Supplementary Information, Table S1. PCR was carried out in a 50 μl reaction mixture containing 100 ng of genomic DNA, 1 U Prime STAR HS DNA polymerase (TAKARA BIO, Ohtsu, Japan), 200 μM dNTPs and 0.2 μM of each primer and amplified for 40 cycles of 98 °C for 10 s, 55 °C for 5 s and 72 °C for 60 s. Amplified products were purified using the MinElute PCR Purification Kit (Qiagen). Sequencing was performed using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on a 3130 Genetic Analyzer (Applied Biosystems) according to the manufacturer's instructions, in both directions. Mutations were confirmed by several independent experiments. For NS2, PCR that used the exon 8 primer set produced two distinctive fragments (data not shown); therefore, we purified both of these fragments separately and sequenced them.
High-density single nucleotide polymorphism microarray mapping analysis
The GeneChip Human Mapping 500K Array Set (Affymetrix, Santa Clara, CA, USA) was used according to the manufacturer's instructions and analyzed using the CNAG (Copy Number Analysis for GeneChip) software.17 Precise deletions were confirmed by analyzing single nucleotide polymorphisms (SNPs) around the predicted breakpoints. The primers used for SNP analyses are listed in Supplementary Information, Table S2. PCR and sequencing were carried out as described above.
Breakpoint isolation and sequencing
Long PCRs were carried out using LA Taq or PrimeSTAR GXL polymerase (TAKARA BIO). The primers used for long PCR are listed in Supplementary Information, Figure S1 and Table S3. Junction fragments were isolated by the PCR-based genomic DNA walking method: Universal GenomeWalker Kit (Clontech, Mountain View, CA, USA). Adaptor ligated genomic libraries were constructed using DNAs from NS8 according to the manufacturer's instructions. Primary amplification was performed using the adaptor primer 1 (AP1) and Z4-0111: 5′-GCTCCAAATTTTGTTCATGGGCATACACT-3′. Secondary amplification was performed using the nested adaptor primer 2 (AP2) and Z1-0214: 5′-ATCAGAAACCTGCATTCAAAAGCACCTG-3′. A Dra I digested library produced a 2.5-kb extra fragment that was isolated and sequenced. The primers used for the confirmation of the deletion in family 5 were as follows: Z4-0111, 5′-GCTCCAAATTTTGTTCATGGGCATACACT-3′; and AF, 5′-TATACCCATTCAGCAATAGATTCCCATTCC-3′.
DNAs of 100 unrelated Japanese individuals were obtained from the Health Service Research Resources Bank (Osaka, Japan) and were tested for the presence of the rearrangement using the primers Z4-0111 and AF.
Results
Sequencing analysis showed that five of the six BCNS families had mutations in PTCH1 coding sequences. One nonsense mutation, one 1-bp deletion, two 2-bp deletions and one 58-bp duplication, all caused premature termination resulting in the truncation of the PTCH1 protein (Table 1). NS1 had a 2-bp deletion at nt239-240 (c.239_240delGA) in exon 2. This frameshift mutation introduces a stop codon at amino acid residue 88 (p.Arg80fs). In NS2, PCR that used the exon 8 primer set produced two distinctive fragments (data not shown). These were purified and sequenced. The results showed that NS2 had a 58-bp duplication at nt1138-1195 (c.1138_1195dup) in exon 8. This duplication produces a stop codon at amino acid residue 399 (p.Trp399X). NS9 and its child (NS10) had a 1 base substitution at nt1196 (c.1196G>A) in exon 8. This causes a Tryptophan to a stop codon substitution at amino acid residue 399 (p.Trp399X). NS3 and its child (NS4) had a 2-bp deletion at nt1670-1671 (c.1670_1671delCA) in exon 12. This causes a frameshift and creates a new stop codon at codon 625 (p.Thr557fs). NS5 had a 1-bp deletion at nt2029 (c.2029delA) in exon 14. This deletion leads to a frameshift and creates a premature stop codon at codon 692 (p.Thr677fs).
Mutations were not detected in patients of family 5 (NS6, its father (NS8) and brother (NS7)) (see Figure 1), within coding exons. However, SNPs in introns indicated the possible deletion of the PTCH1 region in patients of family 5. NS6, NS7 and their mother represent ‘C’ at rs574688, whereas NS8 represents ‘G’ (Table 2). To check for possible large deletions in this region, we carried out a high-density SNP mapping array analysis. This analysis showed an ∼1.2-Mb deletion, including the whole PTCH1 gene, in NS6 (data not shown) and NS8 (Figure 2a) in one allele. The deleted regions were confirmed by sequencing several SNPs around the predicted junction regions. Figure 2b shows the signal intensity of the microarray profile of NS6 and NS8 around the predicted boundaries. NS6 and NS8 had almost similar profiles. To determine more precise junction regions, we analyzed SNPs around the predicted boundaries in members of family 5 (Table 2). The proximal boundary was shown to be between rs11789111 (nn 96066113) and rs11791327 (nn 96070886) corresponding to the first intron of the Znf169 gene. The distal boundary was assigned between rs28455201 (nn 97354024) and rs16909982 (nn 97357272) corresponding to ∼30 kb upstream of the PTCH1 gene transcription initiation site. This deletion includes the ZNF169, FAM22F, HIATL1, FBP2, FBP1, C9orf3, FANCC and PTCH1 genes and three microRNA genes MIRN23B, MIRN27B and MIRN24-1 (Figure 2a).

Microarray analysis of patients of family 5. (a) Microarray profile of NS8 patient shows a 1.2-Mb deletion at 9q22.32. Genes within this predicted deleted region are listed. (b) Signal intensity around the estimated breakpoint of NS8 (red) and NS6 (blue). Arrows show single nucleotide polymorphisms (SNPs) indicated in Table 2. 1: rs9299417, 2: rs3118756, 3: rs16909962 and 4: rs558448.
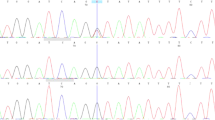
To determine the precise breakpoint sequences, we carried out several long PCRs. However, none of the primer sets produced PCR products (data not shown). Therefore, to analyze the junctions, we tried to clone the breakpoint fragments using a PCR-based genomic DNA walking method (see section Materials and methods). The proximal walking primers produced an additional 2.5-kb fragment using a Dra I digested library. The sequence indicated that the proximal breakpoint is located in an Alu sequence and that the sequence after the breakpoint is also located in an Alu sequence that is ∼0.2 Mb distal from the estimated distal breakpoint in an inverse orientation (Figure 3a and c). The same rearrangement was observed in all BCNS patients of family 5, but not in the unaffected mother and an unrelated BCNS patient (Figure 3b). None of the DNA samples of the 100 unrelated Japanese individuals showed this rearrangement (data not shown). Thus, this rearrangement is specific for patients of family 5. Distal primers failed to produce additional fragments using the GenomeWalker libraries. Although we still do not know the precise structure of the breakpoint at present, these data indicated the complicated rearrangement of this region and explained the failure of long PCRs.

Breakpoint sequence of NS8 and possible mechanism of the rearrangement. (a) Breakpoint sequence is aligned to the wild-type sequences. Alu sequences are shaded. The sites of breakpoints are within the boxed bases. Asterisk (*) corresponds to nn 96070057 of chromosome 9, and dot (•) corresponds to nn 97646373 of chromosome 9 (same as in (c)). (b) PCR confirmation of the rearrangement in patients of family 5;. lane 1: NS8, lane 2: mother, lane 3: NS6, lane 4: NS7 and lane 5: NS9. Primers used for the experiment are P1 (Z4-0111) and P2 (AF) (see panel c). Only patients of family 5 (NS6, 7, 8) produce a 1.6-kb band. (c) Possible mechanism for the rearrangement. Colors indicate the following: orange, proximal from the breakpoint; green, predicted deleted region; blue, inverted region; black, distal from the breakpoint. All bold arrows indicate orientation. Bold red arrow shows the path of the rearrangement. Locations of primers used for PCR analysis in (panel b) are illustrated as black thin arrows (P1, P2).
Discussion
In this study, we identified PTCH1 germline mutations or deletions in all the six Japanese BCNS families. Five families showed small mutations that resulted in protein truncation due to premature termination codons. These patterns of mutation are common in BCNS patients, although the positions vary. According to the publicly available PTCH1 mutation database (http://www.cybergene.se/cgi-bin/w3-msql/ptchbase/index.html), more than 160 PTCH1 mutations in patients with BCNS have been identified. The PTCH1 mutations reported so far in BCNS are scattered over the entire gene without any apparent hot spots.11 All available PTCH1 molecular data of the Japanese BCNS patients also indicated a dispersed distribution of mutations in all regions and suggested no founder effect (Table 3).16, 18, 19, 20, 21, 22, 23, 24, 25, 26 The typical symptoms of the Japanese BCNS patients are characterized by palmar and plantar pits, odontogenic keratocysts and abnormal skeletons and are sometimes accompanied by BCC, especially in elderly patients. On the other hand, BCC is observed at a frequency of up to 97% in Caucasian BCNS patients with the same symptoms as the Japanese.2, 3 A low frequency of BCC onset was also reported in African-American patients.3 The differences of type and onset of symptoms in BCNS in different ethnic populations clearly showed the participation of other factor(s) besides PTCH1 in BCNS phenotypes. A follow-up survey and comparative analyses between the Japanese and Caucasian BCNS patients will elucidate the mechanism of onset of tumors in BCNS patients.
With direct sequencing of all exons, mutations are still unidentified in NS6, 7, 8 (family 5). Phenotypically, patients of family 5 showed very similar phenotypes to the others, except that NS7 had XXY chromosomes. In approximately 10–50% of BCNS cases, mutations were not observed in the PTCH1 coding sequences. Recently, a SNP-based copy number array was applied successfully in several analyses to detect copy numbers of the whole genome.27 Fujii et al.28 applied this method to BCNS cases with PTCH1 mutations that were not previously identified and found deletions with the PTCH1 gene. They also successfully determined their breakpoints. We have carried out the SNP-based copy number analysis and found that NS6 and NS8 had a deletion of ∼1.2 Mb at 9q22.32 (Figure 2a). This deletion contained at least eight coding genes and three microRNA genes, including PTCH1. Patients of family 5 showed no strange phenotypes. Therefore, allelic loss of these genes, except PTCH1, might not influence any observable phenotypes in family 5 at present. Apart from PTCH1, only PTCH2, the second homolog of Drosophila ptc gene, has been identified as a gene responsible for a Chinese BCNS family.29 Thus, application of the microarray-based copy number analysis is a high priority to identify PTCH1 gene alterations in BCNS patients that are not formally identified by exon sequencing.
In addition to the deletion, the sequence analysis of the breakpoint in family 5 showed an inversion between two Alu elements located in the same direction. The involvement of Alu elements in recombination is reported as one of the causes of human genetic diseases, including tumors.30, 31 Although the precise structure of this region is still unknown, a possible mechanism of the large deletion and inversion is illustrated in Figure 3c. Application of a high-density SNP mapping microarray analysis enables us to detect deletions and amplifications at a genome-wide level. This analysis also enabled us to confirm the breakpoint sequences relatively easily. Accumulation of the deletion and/or amplification data will help to search for genetic factors involved in the BCNS phenotypes and the basic mechanisms for gene rearrangements.
References
Gorlin, R. J. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 66, 98–113 (1987).
Evans, D. G., Ladusans, E. J., Rimmer, S., Burnell, L. D., Thakker, N. & Farndon, P. A. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J. Med. Genet. 30, 460–464 (1993).
Kimonis, V. E., Goldstein, A. M., Pastakia, B., Yang, M. L., Kase, R., DiGiovanna, J. J. et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am. J. Med. Genet. 69, 299–308 (1997).
Hahn, H., Wicking, C., Zaphiropoulous, P. G., Gailani, M. R., Shanley, S., Chidambaram, A. et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85, 841–851 (1996).
Johnson, R. L., Rothman, A. L., Xie, J., Goodrich, L. V., Bare, J. W., Bonifas, J. M. et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272, 1668–1671 (1996).
Ingham, P. W. & McMahon, A. P. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087 (2001).
McMahon, A. P., Ingham, P. W. & Tabin, C. J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 53, 1–114 (2003).
Bale, A. E. Hedgehog signaling and human disease. Annu. Rev. Genomics Hum. Genet. 3, 47–65 (2002).
Cohen, M. M. Jr The hedgehog signaling network. Am. J. Med. Genet. 123A, 5–28 (2003).
Beachy, P. A., Karhadkar, S. S. & Berman, D. M. Tissue repair and stem cell renewal in carcinogenesis. Nature 432, 324–331 (2004).
Lindström, E., Shimokawa, T., Toftgård, R. & Zaphiropoulos, P. G. PTCH mutations: distribution and analyses. Hum. Mutat. 27, 215–219 (2006).
Marigo, V., Davey, R. A., Zuo, Y., Cunningham, J. M. & Tabin, C. J. Biochemical evidence that patched is the Hedgehog receptor. Nature 384, 176–179 (1996).
Stone, D. M., Hynes, M., Armanini, M., Swanson, T. A., Gu, Q., Johnson, R. L. et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic Hh. Nature 384, 129–134 (1996).
Evans, D. G., Birch, J. M., Ramsden, R. T., Sharif, S. & Baser, M. E. Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J. Med. Genet. 43, 289–294 (2006).
Wicking, C., Shanley, S., Smyth, I., Gillies, S., Negus, K., Graham, S. et al. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am. J. Hum. Genet. 60, 21–26 (1997).
Fujii, K., Kohno, Y., Sugita, K., Nakamura, M., Moroi, Y., Urabe, K. et al. Mutations in the human homologue of Drosophila patched in Japanese nevoid basal cell carcinoma syndrome patients. Hum. Mutat. 21, 451–452 (2003).
Nannya, Y., Sanada, M., Nakazaki, K., Hosoya, N., Wang, L., Hangaishi, A. et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 65, 6071–6079 (2005).
Minami, M., Urano, Y., Ishigami, T., Tsuda, H., Kusaka, J. & Arase, S. Germline mutations of the PTCH gene in Japanese patients with nevoid basal cell carcinoma syndrome. J. Dermatol. Sci. 27, 21–26 (2001).
Ogata, K., Ikeda, M., Miyoshi, K., Yamamoto, Y., Yamamoto, T., Osaki, T. et al. Naevoid basal cell carcinoma syndrome with a palmer epidermoid cyst, milia and maxillary cysts. Br. J. Dermatol. 145, 508–509 (2001).
Tate, G., Li, M., Suzuki, T. & Mitsuya, T. A new germline mutation of the PTCH gene in a Japanese patient with nevoid basal cell carcinoma syndrome associated with meningioma. Jpn. J. Clin. Oncol. 33, 47–50 (2003).
Nagao, K., Togawa, N., Fujii, K., Uchikawa, H., Kohno, Y., Yamada, M. et al. Detecting tissue-specific alternative splicing and disease-associated aberrant splicing of the PTCH gene with exon junction microarrays. Hum. Mol. Genet. 14, 3379–3388 (2005).
Tanioka, M., Takahashi, K., Kawabata, T., Kosugi, S., Murakami, K., Miyachi, Y. et al. Germline mutations of the PTCH gene in Japanese patients with nevoid basal cell carcinoma syndrome. Arch. Dermatol. Res. 296, 303–308 (2005).
Matsuzawa, N., Nagao, T., Shimozato, K., Niikawa, N. & Yoshiura, K. I. Patched homologue 1 mutations in four Japanese families with basal cell nevus syndrome. J. Clin. Pathol. 59, 1084–1086 (2006).
Tachi, N., Fujii, K., Kimura, M., Seki, K., Hirakai, M. & Miyashita, T. New mutation of the PTCH gene in nevoid basal-cell carcinoma syndrome with west syndrom. Pediatr Neurol. 37, 363–365 (2007).
Honma, M., Ohishi, Y., Uehara, J., Ibe, M., Kinouchi, M., Ishida-Yamamoto, A. et al. A novel PTCH1 mutation in a patient of nevoid basal cell carcinoma syndrome. J. Dermatol. Sci. 50, 73–75 (2008).
Otsubo, S., Honma, M., Asano, K., Takahashi, H. & Iizuka, H. A novel germ-line mutation of PTCH1 gene in a Japanese family of nevoid basal cell carcinoma syndrome: are the palmoplantar pits associated with true basal cell carcinoma? J. Dermatol. Sci. 51, 144–146 (2008).
Vissers, L. E., Veltman, J. A., van Kessel, A. G. & Brunner, H. G. Identification of disease genes by whole genome CGH arrays. Hum. Mol. Genet. 14, R215–223 (2005).
Fujii, K., Ishikawa, S., Uchikawa, H., Komura, D., Shapero, M. H., Shen, F. et al. High-density oligonucleotide array with sub-kilobase resolution reveals breakpoint information of submicroscopic deletions in nevoid basal cell carcinoma syndrome. Hum. Genet. 122, 459–466 (2007).
Fan, Z., Li, J., Du, J., Zhang, H., Shen, Y., Wang, C. Y. et al. A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J. Med. Genet. 45, 303–308 (2008).
Batzer, M. A. & Deininger, P. L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 3, 370–379 (2002).
Kolomietz, E., Meyn, M. S., Pandita, A. & Squire, J. A. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 35, 97–112 (2002).
Acknowledgements
We thank Dr Takada for data management and collection, Ms Iemoto for her technical assistance and the Joint-Use Research facilities at Hyogo College of Medicine. This work was supported by a grant-in-aid from the Project for Frontier of Science from the Ministry of Education, Science, Sports and Culture, Government of Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Rights and permissions
About this article
Cite this article
Takahashi, C., Kanazawa, N., Yoshikawa, Y. et al. Germline PTCH1 mutations in Japanese basal cell nevus syndrome patients. J Hum Genet 54, 403–408 (2009). https://doi.org/10.1038/jhg.2009.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.55
Keywords
This article is cited by
-
Nevoid basal cell carcinoma syndrome with a unilateral giant ovarian fibroma in a Japanese 6-year-old girl
European Journal of Pediatrics (2014)
-
Novel PTCH1 mutations in Japanese Nevoid basal cell carcinoma syndrome patients: two familial and three sporadic cases including the first Japanese patient with medulloblastoma
Journal of Human Genetics (2011)
