
Abstract
Elevated erythrocyte destruction in sickle cell disease (SCD) results in chronic hyperbilirubinaemia and, in a subset of patients, cholelithiasis occurs. We investigated whether the (TA)n promoter polymorphism in the UDP-glucuronosyltransferase 1A1 gene (UGT1A1) may modify bilirubin metabolism, influencing bilirubinaemia, predisposition to cholelithiasis and subsequent cholecystectomy, in a group of 153 young SCD patients (mean age 12.0 ± 9.0 years) predominantly of Bantu beta S haplotype. The concomitant effect of alpha thalassaemia was also analysed. Among the several UGT1A1 genotypes found, the most frequent were the (TA)6/(TA)6 (n = 37), (TA)6/(TA)7 (n = 60) and (TA)7/(TA)7 (n = 29). These groups of patients did not significantly differ in age, gender ratio and haemoglobin, foetal haemoglobin and reticulocyte levels. On the other hand, total bilirubin levels were significantly different between groups, with an increased (TA) repeat number being associated with higher bilirubinaemia. Furthermore, both cholelithiasis and cholecystectomy were more frequent in groups with higher (TA) repeat number, although the former association was not statistically significant. None of the mentioned parameters is statistically different within UGT1A1 groups with the presence of alpha thalassaemia. Thus, the UGT1A1 promoter polymorphism may represent an important nonglobin genetic modifier of Bantu SCD patients’ clinical manifestations, even at a young age.
Similar content being viewed by others

Introduction
Sickle cell disease (SCD; OMIM #603903) is the most common genetic abnormality affecting people of African ancestry. It is caused by a molecular defect in the haemoglobin (Hb) beta (β)-chain, where valine is substituted for glutamic acid in the sixth amino acid position. The resulting variant is called Hb S as opposed to the normal adult Hb A. The polymerisation of deoxygenated Hb S is the primary event in the molecular pathogenesis of SCD, resulting in a distortion of the red cell shape and a marked decrease in its deformability. SCD is characterised by a chronic haemolytic anaemia and recurrent vasoocclusive episodes, leading to multisystemic complications with a wide variation in severity (Serjeant 2001). Part of this clinical heterogeneity can be explained by the coinheritance of genetic modifiers linked or nonlinked to the globin gene clusters (Steinberg and Adewoye 2006).
It is known that the accelerated erythrocyte destruction in SCD often leads to chronic hyperbilirubinaemia, as bilirubin catabolism is the final step in the breakdown of haem from haemoglobin. Consequently, some clinical complications, such as cholelithiasis, recurrent painful abdominal events and cholecystitis are frequent in this pathology.
The primary bilirubin-catabolising hepatic enzyme, UDP-glucuronosyltransferase 1A1 (UGT1A1; Chr 2q37; EC 2.4.1.17), mediates the conjugation of bilirubin into a water-soluble conjugated form that is excreted in the bile. The UGT1A1 promoter contains an A(TA)nTAA sequence with a common (TA)6 tandem repeat. The homozygosity for the (TA)7 repeat has been associated with decreased enzyme function and Gilbert’s syndrome of unconjugated hyperbilirubinaemia (Monaghan et al 1996). Other alleles, a shorter (TA)5 and a longer (TA)8, have been described in people of African descent (Beutler et al. 1998). Cholelithiasis, by promoting cholecystitis, is responsible for high levels of morbidity in SCD patients, and elective cholecystectomy is therefore recommended for patients developing this complication.
The aim of this study was to determine whether the promoter UGT1A1 polymorphism (resulting in UGT1A1 activity variation) could modify bilirubin metabolism and affect serum bilirubin concentration in young SCD patients, thereby influencing the development of cholelithiasis and subsequently cholecystectomy. Also, the effect of alpha thalassaemia (α-thalassaemia) within UGT1A1 genotype groups was investigated.
Patients and methods
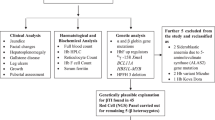
This study was performed on 153 young Portuguese SCD patients (mean age 12.0 ± 9.0 years), the majority descending from African immigrants (preponderantly from Angola, Sao Tome and Principe and Cape Verde). All patients, or their legal representatives, gave informed consent prior to their inclusion in the study. SCD steady-state patient data, concerning haemoglobin, foetal haemoglobin, reticulocytes and total bilirubin levels (determined by standard procedures) were averaged for each patient, and clinical information was gathered for cholelithiasis (detected by liver/biliary ultrasound scans) and cholecystectomy. Genomic DNA was extracted from peripheral blood leukocytes by a salting-out procedure (Miller et al 1988). The homozygosity for the SCD mutation was confirmed by a bidirectional allele-specific polymerase chain reaction (PCR) as described (Liu et al. 1997). The number of (TA) repeats in the UGT1A1 promoter was analysed by a PCR-based methodology using the primers described by Monaghan et al. (1996) in which the sense primer was 5-carboxyfluorescein (FAM) fluorescent-tagged. One aliquot of each PCR product was analysed by Gene Scan 3.7 software, along with a molecular weight marker.
Beta globin (β-globin) gene-cluster haplotypes were assigned after examining nine restriction endonuclease sites within the cluster [Hinc II (ε), Xmn I (5′ to Gγ), Hind III (within Gγ and Aγ), Hinc II (within and 3′ to ψβ), Hinf I (5′ to β), Ava II (within β), and Hinf I (3’ to β)]. Aliquots of the amplified products were digested with the appropriate restriction enzymes under the conditions recommended by the manufacturer. Haplotypes were established by determining the presence or absence of cleavage at each site and by compiling the results into one pattern classified according to Orkin et al. (1982). The allelic phase was determined by family studies whenever necessary. The −α3.7 kb deletion was searched for by gap PCR, as previously described (Dodé et al. 1992). Individuals were scored according to the presence/absence of deletional alleles. In order to perform statistical analyses, SCD patients presenting one or two deletional (−α3.7) alleles were integrated in the same group, named α-thal (see Table 3).
All statistical analyses were performed using the statistical software package SPSS 14.0. The significance of the differences between groups of patients was assessed using the Kruskal–Wallis, chi-square, analysis of variance (ANOVA), Wald, Mann–Whitney U or Fisher’s exact tests, as appropriate. The odds ratios (OR) were obtained by applying a logistic regression model using the UGT1A1 genotype as the independent variable and the cholelithiasis and cholecystectomy as dependent variables. The reference class was the 6/6 UGT1A1 genotype. Significance for statistical analysis was defined as a P < 0.05.
Results
The homozygosity for the sickle cell mutation (HBB:c.20A > T) was confirmed in all patients. The UGT1A1 PCR fragments obtained ranged between 96 and 102 bp, which corresponded to a (TA)5–8 variation. Consequently, several UGT1A1 genotypes were found: 5/5 (n = 1); 5/6 (n = 9); 5/7 (n = 5); 5/8 (n = 3); 6/6 (n = 37); 6/7 (n = 60); 6/8 (n = 6); 7/7 (n = 29) and (7/8) (n = 3); (Appendix 1 of Supplementary Material). Only patients belonging to the three largest genotype groups (namely, 6/6, 6/7 and 7/7) were considered for statistical analyses. Comparisons performed for these three groups of patients did not reveal any significant difference concerning age, gender ratio, and Hb, Hb F and reticulocyte levels, which denotes a similar degree of haemolysis between groups. However, total bilirubin levels were significantly different between groups (P = 0.003; Kruskal–Wallis test), with an increased (TA) repeat number being associated with higher bilirubinaemia (Table 1).
The chi-square tests used for cholelithiasis and cholecystectomy data showed that both cholelithiasis (P = 0.407) and cholecystectomy (P = 0.016) were increased in the patients with higher (TA) repeat number, although the former difference was not statistically significant (Table 1). The OR of having cholelithiasis for genotypes 6/7 [OR = 1.500; 95% confidence interval (CI) = 0.595–3.784) and 7/7 (OR = 2.057; 95% CI = 0.708–5.980] was increased when compared with 6/6. The same comparison for cholecystectomy revealed an increased risk only for genotype 7/7 (OR = 4.714; 95% CI = 0.869–25.581) (Table 2).
To gain further insight into the modifying effect of UGT1A1 polymorphism, the βS haplotype and the α-globin genotype were determined in our SCD patients. The prevalence of the Bantu haplotype was similar among the three UGT1A1 groups (Bantu allelic frequency in the 6/6 UGT1A1 group was 73.0%; in the 6/7 group 62.3%; and in the 7/7 group 67.2%; P = 0.305). The overall α-thalassaemia (−α3.7) allelic frequency was 26% and was not significantly different between UGT1A1 genotype groups. We found that in young SCD patients predominantly of Bantu βS haplotype, the coinheritance of α-thalassaemia did not significantly influence the level, total bilirubin level, cholelithogenesis and the need for cholecystectomy (Table 3).
Phenotype features of patients belonging to the less frequent genotype groups are summarised in Appendix 1 of Supplementary Material.
Discussion
In this study, we investigated the genetic architecture of a young population with a SCD subphenotype characterised by hyperbilirubinaemia and cholelithiasis. The (TA) repeat number found within the UGT1A1 gene promoter varied between 5 and 8, which was expected due to the African ancestry of this population (Beutler et al. 1998).
The coinheritance of Gilbert’s syndrome determinant and genetic alterations associated with disorders that increase turnover of red blood cells or their precursors has been reported to elevate bilirubin levels, e.g. in β-thalassaemia (Galanello et al. 1997, 1999b), G6PD deficiency (Galanello et al. 1999a) and SCD (Adekile et al. 2005; Chaar et al. 2005; Fertrin et al. 2003; Harverfield et al. 2005; Passon et al. 2001). In fact, in addition to the results presented here, the mentioned studies performed in SCD patients have shown that homozygosity for the (TA)7UGT1A1 genotype significantly influences bilirubin metabolism and is associated with higher steady-state concentrations of unconjugated serum bilirubin. At least in our case, this fact could not be explained by increased haemolysis, as there are no significant differences in average Hb concentration and in reticulocyte count among the different UGT1A1 genotype groups (Table 1). Some other studies were performed regarding the influence of the UGT1A1 polymorphism on cholelithiasis, but the association has been less consistent that with a more proximal phenotype, namely, hyperbilirubinaemia. Moreover, age has been considered a modifier of UGT1A1 polymorphism influence. As an example, Harverfield et al. (2005) observed that the (TA)7 homozygosity is a significant risk factor for cholelithiasis development only in older SCD patients (34.6 ± 10.9 years) but not in a younger adult cohort group (25.0 ± 2.9 years). Although gallstones were identified at a mean age of 15.1 years in 40.6% of this cohort group, there was no association with UGT1A1 (TA)n genotypes (Harverfield et al. 2005). On the other hand, Fertrin et al. (2003) analysed a group of adult SCD patients (28.5 ± 10.7 years) and observed that although the frequency of cholelithiasis was higher in patients with the 7/7 genotype, the difference was not statistically significant (Fertrin et al. 2003). Considering the need for cholecystectomy, the difference between 6/7 and 6/6 patients was statistically significant, although the difference between the other groups was not (Fertrin et al. 2003). Furthermore, in studies performed in SCD children (3–19 years) the development of symptomatic cholelithiasis requiring cholecystectomy was significantly higher for children with the 7/7 genotype than those with the 6/7 or 6/6 genotypes (Passon et al. 2001). Finally, Chaar et al. (2005) observed that the frequency of cholelithiasis was significantly higher in an SCD group of children having 7/7 or 7/8 genotype. On the whole, analysing our results along with previously published data, we can conclude that the UGT1A1 promoter genotype is unquestionably an important genetic determinant of hyperbilirubinaemia in SCD. However, its influence on cholelithiasis development is not so straightforward and seems to be modulated by other factors, among which age is relevant. Concerning patients who need cholecystectomy, their inclusion in the 7/7 genotype group is remarkable, thereby confirming the penetrance of this genotype.
It is known that the coinheritance of α-thalassaemia and SCD determinants reduces the mean corpuscular haemoglobin concentration of red blood cells, which tends to inhibit sickling. It was observed that the coexistence of α-thalassaemia decreases the chance of developing gallstones in Arab Indian haplotype SCD patients, which may be related to lower haemolysis, as suggested by their predisposition to present, in general, higher mean Hb levels (Haider et al. 1998). In addition, Adekile et al. (2005) observed that the β-globin haplotype and the coexisting α-thalassaemia did not have significant influence on serum bilirubin levels in Arab Indian haplotype SCD patients (Adekile et al. 2005). Furthermore, Chaar et al. (2006) showed that although α-thalassaemia is associated with a modest reduction in haemolysis and unconjugated bilirubin level, UGT1A1 polymorphism outweighs its effect on cholelithogenesis in SCD patients predominantly of Benim βS haplotype (Chaar et al. 2006). Vasavda et al. (2007) also observed that α-thalassaemia was significantly associated with reduced bilirubin levels in SCD adult patients. In our young SCD patient series predominantly of Bantu βS haplotype, we found that the coinheritance of α-thalassaemia does not significantly influence Hb level, total bilirubin level, cholelithogenesis and the need for cholecystectomy (Table 3). However, we observed that the coinheritance of α-thalassaemia is slightly associated with a positive effect on the mentioned parameters, mainly in the 7/7 genotype group in which patients presented higher haemoglobin level, lower bilirubin, fewer cholelithiasis and undergo cholecystectomy less frequently.
In conclusion, our study, the first performed regarding young Bantu SCD patients, reinforces the concept that the sickle cell mutation alone is not sufficient to explain the wide range of phenotypic expression characteristic of SCD. Our results attribute to UGT1A1 promoter polymorphism an important modifying effect of the clinical manifestations in Bantu SCD patients, namely, in bilirubin metabolism, cholelithiasis development and the need for cholecystectomy, even at a young age. Also, we observed that in these patients, the modulating effect of the UGT1A1 promoter polymorphism is not significantly altered by the coinheritance of α-thalassaemia determinants.
References
Adekile A, Kutlar F, McKie A, Addington A, Elam D, Holley L, Clair B, Kutlar A (2005) The influence of uridine diphosphate glucuronyl transferase 1A promoter polymorphisms, βS-globin trait and HbF on steady-state bilirubin levels in sickle cell anemia. Eur J Haematol 75:150–155
Beutler E, Gelbart T, Demina A (1998) Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA 95:8170–8174
Chaar V, Kéclard L, Diara JP, Leturdu C, Elion J, Krishnamoorthy R, Clayton J, Romana M (2005) Association of UGT1A1 polymorphism with prevalence and age at onset of cholelithiasis in sickle cell anemia. Haematologica 90:188–193
Chaar V, Keclard L, Etienne-Julan M, Diara JP, Elion J, Krishnamoorthy R, Romana M (2006) UGT1A1 polymorphism outweighs the modest effect of deletional (−3.7 kb) alpha-thalassemia on cholelithogenesis in sickle cell anemia. Am J Hematol 81:377–379
Dodé C, Krishnamoorthy R, Lamb J, Rochette J (1992) Rapid analysis of α3.7 thalassaemia and αααanti3.7 triplication by enzymatic amplification analysis. Br J Haematol 83:105–111
Fertrin KY, Melo MB, Assis AM, Saad STO, Costa FF (2003) UDP- glucuronosyltransferase 1 gene promoter polymorphism is associated with increased serum bilirubin levels and cholecystectomy in patients with sickle cell anemia. Clin Genet 64:160–162
Galanello R, Perseu L, Melis M, Cipollina L, Barella S, Giagu N (1997) Hyperbilirubinaemia in heterozygous beta-thalassaemia is related to co-inherited Gilbert’s syndrome. Br J Haematol 99:433–436
Galanello R, Cipollina M, Carboni G, Perseu L, Barella S, Corrias A, Cao A (1999a) Hyperbilirubinemia, glucose-6-phosphate dehydrogenase deficiency and Gilbert’s syndrome. Eur J Pediatr 158:914–916
Galanello R, Cipollina M, Dessi C, Giaqu N, Lai E, Cao A (1999b) Co-inherited Gilbert’s syndrome: a factor determining hyperbilirubinemia in homozygous beta-thalassemia. Haematologica 84:103–105
Haider MZ, Ashebu S, Aduh P, Adekile AD (1998) Influence of alpha-thalassemia on cholelithiasis in SS patients with elevated Hb F. Acta Haematol 100:147–150
Harverfield EV, McKenzie CA, Forrester T, Bouzekri N, Harding R, Serjeant G, Walker T, Peto TEA, Ward R, Weatherall DJ (2005) UGT1A1 variation and gallstone formation in sickle cell disease. Blood 105:968–972
Liu Q, Thorland EC, Heit JA, Sommer SS (1997) Overlapping PCR for bidirectional PCR amplification of specific alleles: a rapid one-tube method for simultaneously differentiating homozygotes and heterozygotes. Genome Res 7:389–398
Miller SA, Dykes DD, Polesky HF (1988) A simple salting-out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Monaghan G, Ryan M, Seddon R, Hume R, Burchell B (1996) Genetic variation in bilirubin UDP-glucuronosyltransferase gene promoter and Gilbert’s syndrome. Lancet 347:578–581
Orkin SH, Kazazian HH, Antonarakis SE, Goff SC, Boeham CD, Sexton JP, Waber PG, Giardina PJ (1982) Linkage of β-thalassaemia mutations and β-globin gene polymorphisms with DNA polymorphism in human β-globin gene cluster. Nature 296:627–631
Passon RG, Howard TA, Zimmerman SA, Schultz WH, Ware RE (2001) Influence of Bilirubin Uridine Diphosphate-Glucuronosyltransferase 1A promoter polymorphisms on serum bilirubin levels and cholelithiasis in children with sickle cell anemia. J Pediatr Hematol Oncol 23:448–451
Serjeant GR (2001) The emerging understanding of sickle cell disease. Br J Haematol 112:3–18
Steinberg MH, Adewoye AH (2006) Modifier genes and sickle cell anaemia. Curr Opin Hematol 13:131–136
Vasavda N, Menzel S, Kondaveeti S, Maytham E, Awogbade M, Bannister S, Cunningham J, Eichholz A, Daniel Y, Okpala I, Fulford T, Thein SW (2007) The linear effects of α-thalassaemia, the UGT1A1 and HMOX1 polymorphisms on cholelithiasis in sickle cell disease. Br J Haematol 138:263–270
Acknowledgments
We thank the “Unidade Laboratorial de Utilização Comum” of the National Institute of Health Dr. Ricardo Jorge for technical support. This work was partially supported by FCT grant IME/MGI/49853/2003, “Programa de Financiamento Plurianual do CIGMH” and “Programa Operacional Saúde – Saúde XXI”. The experiments performed in our research work comply with the current Portuguese applicable laws. This work obtained the approval of the local ethics committee.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Martins, R., Morais, A., Dias, A. et al. Early modification of sickle cell disease clinical course by UDP-glucuronosyltransferase 1A1 gene promoter polymorphism. J Hum Genet 53, 524–528 (2008). https://doi.org/10.1007/s10038-008-0281-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-008-0281-3
Keywords
This article is cited by
-
Influence of UGT1A1 promoter polymorphism, α-thalassemia and βs haplotype in bilirubin levels and cholelithiasis in a large sickle cell anemia cohort
Annals of Hematology (2021)
-
Uridine diphosphate glucuronosyl transferase 1A (UGT1A1) promoter polymorphism in young patients with sickle cell anaemia: report of the first cohort study from Nigeria
BMC Medical Genetics (2019)
