
Abstract
Eight mutations of the α subunit of β-hexosaminidase A gene (HEXA) were identified in eight patients with GM2 gangliosidosis variant B. They were five missense mutations, two splice-site mutations, and one two-base deletion. Five of them, R252L (CGT→CTT), N295S (AAT→AAC), W420C (TGG→TGT), IVS 13, +2A→C, and del 265–266AC (exon 2), were novel mutations responsible for infantile acute form of GM2 gangliosidosis. Two missense mutations, R499H and R499C, were found in one allele of two patients with attenuated phenotypes. The patient with R499C showed a late infantile form, and the other patient with R499H showed a juvenile form. These two mutations have been reported previously in the patients of other ethnic groups, and they have been known to cause attenuated phenotypes. The milder phenotypes of GM2 gangliosidosis variant B, different from the infantile acute form, have not been reported so far in Japan, and this is the first report of Japanese patients with attenuated phenotypes and their molecular analysis.
Similar content being viewed by others


Introduction
Tay-Sachs disease, GM2 gangliosidosis variant B, is an autosomal recessive disorder primarily affecting the central nervous system. It is caused by mutations in the gene encoding the α subunit of β-hexosaminidase A (Hex A), a lysosomal enzyme composed of α and β polypeptides. Hex A requires the assistance of GM2 activator protein for hydrolysis of the lipophilic ganglioside GM2 in the hydrophilic environment of the lysosome. Mutations in the β subunit and GM2 activator protein respectively result in the two other GM2 gangliosidoses known as Sandhoff disease (variant O) and activator protein deficiency (variant AB). Deficient catabolism and abnormal accumulation of ganglioside GM2 is a common feature in all GM2 gangliosidoses, and consequently they all exhibit similar clinical symptoms.
About hundred mutations in HEXA have been described in the literature (Gravel et al., 2001) so far. Some mutations are commonly found in ethnically or geographically isolated populations such as Ashkenazi Jewish patients (Myerowitz and Costigan, 1988; Arpaia et al., 1988; Myerowitz, 1988; Ohno and Suzuki, 1988) or French Canadian patients (Myerowitz and Hogikyan, 1986). We previously reported two common mutations among Japanese patients with Tay-Sachs disease. One was IVS 5, -1 G→T accounting for 80% of the mutant alleles (Tanaka et al., 1993) and the other was del nt613C accounting for 5% of the mutant alleles (Tanaka et al., 1999). Since both mutations result in null alleles and are responsible for infantile acute form of GM2 gangliosidosis, patients who show other clinical phenotypes than infantile acute form have not been reported so far in the Japanese population.
We report here two patients with different attenuated clinical phenotypes of GM2 gangliosidosis caused by the different missense mutations in the same codon of HEXA. This is the first report of Japanese patients with attenuated phenotypes other than infantile acute form. In addition, we report five novel mutations responsible for infantile acute form of GM2 gangliosidosis variant B.
Materials and methods
Molecular and biochemical analysis
Cultured skin fibroblasts obtained from the patients with GM2 gangliosidosis variant B were used for the study in patients 1–6. Peripheral lymphocytes and chorionic villi were used in patients 7 and 8, respectively.
Genomic DNA was extracted from cells or tissue by the standard method. In the first step, two common mutations (IVS 5, -1 G→T and del nt613C) were screened by the previously reported method (Tanaka et al., 1999), and eight patients (patients 1–8) who had unknown mutations were selected. In the next step, PCRs and the sequencing were performed as described previously (Tanaka et al., 1999) to analyze the unknown mutations.
Enzyme analysis was carried out in the tissue homogenates with the substrates of 4-methylumbelliferyl-N-acetyl β-glucosaminide for the activity of hexosaminidase A plus B and 4-methylumbelliferyl-N-acetyl β-glucosamine-6-sulfate for the specific activity of hexosaminidase A according to the method of Suzuki (1987). All the patients gave their informed consent prior to their inclusion in this study.
Patients’ reports
Patients 1, 2, 3, 5, 7, and 8 showed the typical phenotype of infantile acute form of Tay-Sachs disease. The symptoms appeared at around 6 months of age. They never walked and became bed-ridden at about 1 year old. Patients 4 and 6 showed different attenuated phenotypes as described following. All patients had no records of consanguinities.
Patient 4, a 9-year-old boy: The patient developed normally until 1 year old when he could walk and speak one-word sentences. Mother noted that he showed no development after that, and consulted a doctor when he was 1 year and 6 months. The doctor found that he had hyperacusis and a cherry-red spot on the macula, and the diagnosis of GM2 gangliosidosis was made. He could not stand at age 2, and clonic seizures appeared at age 3. Nasal-tube feeding was introduced when he was 3. He was bed-ridden, had no social communications, and showed some myoclonic movements at the age of 7 years.
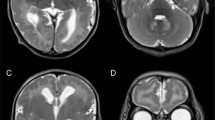
Patient 6, an 8-year-old girl: She developed normally until 3 years of age. She showed dysphemia at 3 years and 9 months. Then, she gradually lost words. She had the first attack of generalized seizure at 5 years and 10 months. Her EEG showed spike discharges at both temporal regions. The seizures appeared several times a day, and they were intractable by oral administration of anticonvulsants. She showed some difficulty with communication at 6 years and 5 months, nystagmus at 7 years and 1 month, and could not walk at 7 years and 2 months. At 7 years and 10 months, she was bed-ridden and showed some chorea-like involuntary movements. She could neither move nor communicate socially. She had no macular cherry-red spot. The brain MRI showed severe cortical atrophy. The diagnosis of GM2 gangliosidosis variant B was made by enzyme analysis.
Results
The eight patients, who had unknown mutations in either of the two alleles, were selected by the screening of the two common mutations, as described previously. These patients were examined enzymatically and molecularly, and the results were summarized in Table 1. All except patient 1 had IVS 5, -1 G→T in one allele. Eight mutations were identified for 8 patients , which were five missense mutations, two splice-site mutations, and one two-base deletion. Five, R252L (CGT→CTT), N295S (AAT→AAC), W420C (TGG→TGT), IVS 13, +2A→C, and del 265–266AC (exon 2), were novel mutations responsible for infantile acute form of GM2 gangliosidosis.
Two different missense mutations were found in the same codon in two patients (patients 4 and 6), R449C (CGT→TGT) and R449H (CGT→CAT), which caused the attenuated clinical phenotypes. Both mutations were generated at CpG spot. Patient 4 had R449C and showed late infantile form, and patient 6 had R449H and showed juvenile form. Hex A activity in these patients seemed to be higher than in other patients with infantile acute form, as shown in Table 1.
Discussion
A huge number of mutations of HEXA gene have been reported previously. Most of them, about 90%, were known to cause the infantile acute form, so called Tay-Sachs disease, whose clinical phenotype is almost similar among patients from different ethnic groups. However, the clinical phenotypes of the chronic form are very heterogeneous (Gravel et al., 2001). Sometimes they show different phenotypes with the same mutation. The reason of this phenomenon is still unclear. Interactions between the mutant proteins from both alleles and the balance of those proteins must play an important role, which might be different among individuals even though they have the same mutations. Moreover, unknown effect from other genes or other proteins might occur.
GM2 gangliosidosis variant B in Japan is very unique. Most patients show infantile acute clinical phenotype and have the same mutation of IVS 5, -1G→T, which is considered to originate in Japan (Tanaka et al., 1994). Moreover, patients with milder forms have not been reported so far in the Japanese population.
We reported two Japanese patients with different attenuated phenotypes (late infantile and juvenile forms) and identified the disease-causing mutations in each patient. Both had the mutation of IVS 5, -1G→T in one allele. The mutations in another allele were R499C for one patient (patient 4) and R499H for the other (patient 6). Patient 4 showed a late infantile form, which was more severe than patient 6 but milder than other typical Japanese patients with GM2 gangliosidosis. Patient 6 showed a juvenile form.
Previously, R499C was reported in two patients—a French patient and an Italian patient—with infantile form (Akli et al., 1993), and one patient (Slavic/Irish) with adult form (Mules et al., 1992). R499H was reported in a Scottish/Irish patient (Paw et al., 1990) and a Scottish/English patient (Triggs-Raine et al., 1991) with juvenile form, and in a Dutch patient with adult form (Akli et al., 1993). All four, the two R499C patients with infantile form (a French patient and an Italian patient) and the two R499H patients with juvenile form (a Scottish/Irish patient and a Scottish/English patient), had the same null mutation of 4-base insertion in exon 11 in another allele. The molecular situation of the two patients in this report, who had R499C/H and a null mutation (IVS 5, -1G→T), is the same as these four patients with R499C/H and 4-base insertion. Patient 4 (R499C/IVS 5, -1G→T) showed less severe phenotype than the French and the Italian patients (R499C/4-base insertion). Patient 6 (R499H/IVS 5, -1G→T) showed a similar phenotype with the Scottish/Irish and the Scottish/English patients (R499H/4-base insertion).
The reason R499H causes mild phenotypes was previously studied biochemically (Paw et al., 1990). As the α-chain generated from the mutant allele with R499H would not be processed to the mature form, it would be unstable and unable to exit endoplasmic reticulum (ER). Some intracellular milieu might help to exit ER and to reach into lysosomes and create a small amount of mature polypeptide. In R499C, the cysteine residue might create an illegitimate disulfide bridge in the protein with a resultant disruption of the normal three-dimensional structure, causing a more severe clinical phenotype than in R499H (Akli et al., 1993). Our study of the structural analysis by molecular modeling software showed no drastic structural changes in α subunit polypeptides with R499C/H (Matsuzawa et al., 2003), which was consistent with the mild clinical phenotypes.
We also found six mutations in Japanese patients with infantile acute form. One, IVS 6, +1G→A, was reported previously in an American patient with subacute form (Akli et al., 1993). The remaining five mutations were novel. Two-base deletion (del 265, 266AC) would cause a flame shift and generate a stop signal at the 16th codon downstream to make a truncated polypeptide without biological function. As IVS 13, +2 A→C abolished the consensus sequence for the splicing, the mutation would cause impairment of normal splicing. W420C was reported previously in an Irish/German patient with infantile acute form. These two patients, this Irish/German patient and the patient in this report, had different nucleotide substitutions resulting in the same amino acid substitution, TGG to TGC and to TGT, respectively. R252L must be a disease-causing mutation because a different mutation in the same codon (R252H) was previously reported in a Portuguese patient (Ribeiro et al., 1996) who has mutation of B1 variant (a mutation for mild form) in another allele, and the amino acid R255 would be important for the protein structure. As N295 is one of the N-linked glycosylation sites of the α-chain polypeptide (Myerowitz et al., 1985), the amino acid substitution of N295S must cause a significant conformation change of the protein, which was consistent with the severe clinical phenotype of this patient.
References
Akli S, Chomel J-C, Lacorte J-M, Bachner L, Poenaru L, Kahn A (1993) Ten novel mutations in the HEXA gene in non-Jewish Tay-Sachs patients. Hum Mol Genet 2:61–67
Arpaia E, Dumbrille-Ross A, Maler T, Neote K, Tropk M, Troxel C, Stirling JL, Pitts JS, Bapat B, Lamhonwah AM, Mahuran DJ, Schuster SM, Clarke JTR, Lowden JA, Gravel RA (1988) Identification of an altered splice site in Ashkenazi Tay-Sachs disease. Nature 333:85–86
Gravel RA, Kaback MM, Proia RL, Sandhoff K, Suzuki K, Suzuki Kuni (2001) The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds, the Metabolic and molecular bases of inherited disease, 8th edn. McGraw-Hill: New York, pp 3827–3876
Matsuzawa F, Aikawa S, Sakuraba H, Tanaka A, Hoang TNL, Ohno K, Ninomiya H, Sugimoto Y, Doi H (2003) Structural basis of the GM2 gangliosidosis B variant. J Hum Genet (in press) DOI 10.1007/s10038-003-0082-7
Mules EH, Hayflick S, Miller CS, Reynolds LW, Thomas GH (1992) Six novel deleterious and three neutral mutations in the gene encoding the α-subunit of hexosaminidase A in non-Jewish individuals. Am J Hum Genet 50:834–841
Myerowitz R (1988) Splice junction mutation in some Ashkenazi Jews with Tay-Sachs disease: Evidence against a single defect within this ethnic group. Proc Natl Acad Sci USA 85:3955–3959
Myerowitz R, Costigan FC (1988) The major defect in Ashkenazi Jews with Tay-Sachs disease is an insertion in the gene for the α-chain of β-hexosaminidase. J Biol Chem 263:18587–18589
Myerowitz R, Hogikyan ND (1986) A deletion involving alu sequences in the β-hexosaminidase α-chain gene of French Canadians with Tay-Sachs disease. J Biol Chem 262:15396–15399
Myerowitz R, Piekarz R, Neufeld EF, Shows TB, Suzuki K (1985) Human β-hexosaminidase α chain: Cloning sequence and homology with the β chain. Proc Natl Acad Sci USA 82:7830-7834
Ohno K, Suzuki K (1988) A splicing defect due to an exon-intron junctional mutation results in abnormal β-hexosaminidase α chain mRNAs in Ashkenazi Jewish patients with Tay-Sachs disease. Biochem Biophys Res Commun 153:463–469
Paw BH, Moskowitz SM, Uhrhammer N, Wright N, kaback MK, Neufeld EF (1990) Juvenile GM2 gangliosidosis caused by substitution of histidine for arginine at position 499 or 504 of the α-subunit of β-hexosaminidase. J Biol Chem 265:9452–9457
Ribeiro MG, Sonin T, Pinto RA, Fontes A, Ribiero H, Pinto E, Palmeira MM, Sa Miranda MC (1996) Clinical, enzymatic, and molecular characterization of a Portuguese family with a chronic form of GM2-gangliosidosis B1 variant. J Med Genet 33:341–343
Suzuki K (1987) Enzymatic diagnosis of sphingolipidosis. Methods Enzymol 138:727–762
Tanaka A, Sakuraba H, Isshiki G, Suzuki K (1993) The major mutation among Japanese patients with infantile Tay-Sachs disease: A G-to-T transversion at the acceptor site of intron 5 of the β-hexosaminidase α gene. Biochem Biophys Res Comm 192:539–546
Tanaka A, Sakazaki H, Murakami H, Isshiki G, Suzuki K (1994) Molecular genetics of Tay-Sachs disease in Japan. J Inher Metab Dis 17:593–600
Tanaka A, Fujimaru M, Choeh K, Isshiki G (1999) Novel mutations including the second most common in Japan in the β-hexosaminidase α subunit gene, and a simple screening of Japanese patients with Tay-Sachs disease. J Hum Genet 44:91–95
Triggs-Raine BL, Akerman BR, Clarke JTR, Gravel RA (1991) Sequence of DNA flanking the exons of the HEXA gene, and identification of mutations in Tay-Sachs disease. Am J Hum Genet 49:1041–1054
Acknowledgements
We thank the following doctors for providing the patients’ materials: Kazuko Sukegawa at Gifu University (patient 1), Hitoshi Sakuraba at the Tokyo Metropolitan Institute of Medical Science (patient 2), Koji Inui at Osaka University (patients 3 and 5), Susumu Katayama (patient 4) and Yukitoshi Yamaguchi (patient 7) at Toho University, Tokyo, and Tsutomu Takahashi at Akita University (patient 8). We also thank Dr. Hitoshi Sakuraba for the structural analysis of the mutant α subunit polypeptides by computer software.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by the Foundation of Osaka Fellowship (AT, 2001) from Osaka City, Japan, and the grant AT-11557060 from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Rights and permissions
About this article
Cite this article
Tanaka, A., Hoang, L.T.N., Nishi, Y. et al. Different attenuated phenotypes of GM2 gangliosidosis variant B in Japanese patients with HEXA mutations at codon 499, and five novel mutations responsible for infantile acute form. J Hum Genet 48, 571–574 (2003). https://doi.org/10.1007/s10038-003-0080-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0080-9
Keywords
This article is cited by
-
In silico analysis of the effects of disease-associated mutations of β-hexosaminidase A in Tay‒Sachs disease
Journal of Genetics (2020)
-
Identification of novel variants in a large cohort of children with Tay–Sachs disease: An initiative of a multicentric task force on lysosomal storage disorders by Government of India
Journal of Human Genetics (2019)
