
Abstract
CO2 release from soil is commonly used to estimate toxicity of various substances on microorganisms. However, the mechanisms underlying persistent CO2 release from soil exposed to toxicants inhibiting microbial respiration, for example, sodium azide (NaN3) or heavy metals (Cd, Hg, Cu), remain unclear. To unravel these mechanisms, NaN3-amended soil was incubated with position-specifically 13C-labeled glucose and 13C was quantified in CO2, bulk soil, microbial biomass and phospholipid fatty acids (PLFAs). High 13C recovery from C-1 in CO2 indicates that glucose was predominantly metabolized via the pentose phosphate pathway irrespective of inhibition. Although NaN3 prevented 13C incorporation into PLFA and decreased total CO2 release, 13C in CO2 increased by 12% compared with control soils due to an increased use of glucose for energy production. The allocation of glucose-derived carbon towards extracellular compounds, demonstrated by a fivefold higher 13C recovery in bulk soil than in microbial biomass, suggests the synthesis of redox active substances for extracellular disposal of electrons to bypass inhibited electron transport chains within the cells. PLFA content doubled within 10 days of inhibition, demonstrating recovery of the microbial community. This growth was largely based on recycling of cost-intensive biomass compounds, for example, alkyl chains, from microbial necromass. The bypass of intracellular toxicity by extracellular electron transport permits the fast recovery of the microbial community. Such efficient strategies to overcome exposure to respiration-inhibiting toxicants may be exclusive to habitats containing redox-sensitive substances. Therefore, the toxic effects of respiration inhibitors on microorganisms are much less intensive in soils than in pure cultures.
Similar content being viewed by others


Introduction
Soil microbes are the primary drivers of soil organic matter (SOM) cycling. However, microbial activity can be altered by human activities such as fertilization, use of pesticides or soil contamination by heavy metals from mines, industrial, agricultural and technological application. The rate and amount of CO2 evolved from soil are used to evaluate the effects of toxicants (for example, heavy metals and other pollutants) on heterotrophic microorganism activity in SOM decomposition (Babich and Stotzky, 1985; ISO 16072). Persistent CO2 release from soils exposed to toxicants (Voroney and Paul, 1984; Trevors, 1996) or contaminated with heavy metals (Bond et al., 1976; Ausmus et al., 1978; Fliessbach et al., 1994) remains unclear. Understanding the mechanisms responsible for CO2 emission under such disturbances is important for predicting the response of nutrient and C cycles to future anthropogenic environmental changes (Schimel, 2013). Knowledge of metabolic pathways through which organic substances are oxidized to CO2 is crucial in unraveling these mechanisms, making metabolic tracing an invaluable tool for identifying alterations in microbial transformation pathways of organic substances under these unfavorable conditions (Scandellari et al., 2009; Dijkstra et al., 2011a; Dippold et al., 2014).
Microorganisms break down complex plant materials such as cellulose to produce easily available water-soluble substances such as glucose, the most abundant monomer in soil, from which about 60–70% is incorporated into cellular compounds while 30–40% is oxidized for energy (Fischer et al., 2007; Gunina and Kuzyakov, 2015). Sufficient amounts of glucose in soil solution can activate microbial metabolism and induce growth (Blagodatskaya and Kuzyakov, 2013; Mau et al., 2015), hence accelerating SOM decomposition. In sterile soil, approximately 98% of glucose remains K2SO4-extractable within 24 h (Bremer and van Kessel, 1990). Glucose lacks physical or chemical interactions with soil due to an absence of charged functional groups or hydrophobic parts (Fischer et al., 2010; Apostel et al., 2015), making it a potent candidate for tracing soil metabolic processes. Furthermore, the use of position-specifically labeled glucose allows the fate of individual molecular positions to be determined (Scandellari et al., 2009; Dijkstra et al., 2011a; Apostel et al., 2015), permitting detailed reconstruction of microbial metabolic pathways and de novo formed products (Dippold and Kuzyakov, 2013). This provides the toolbox required to elucidate the source of CO2 emission under respiration-inhibited conditions and thus to identify the mechanisms to overcome intoxication in contaminated environments.
In this study, microbial respiration was inhibited via NaN3 addition, a potent electron transport chain inhibitor at cytochrome oxidase and catalase (Keilin, 1936), resulting in cell asphyxiation (Winter et al., 2012). Paradoxically, azide addition increases CO2 efflux from soil (Rozycki and Bartha, 1981; Trevors, 1996). This cannot be attributed to errors in CO2 determination caused by formation of volatile hydrazoic (HN3) acid (Rozycki and Bartha, 1981; Trevors, 1996), as chloroform fumigation also results in persistent CO2 emission (Voroney and Paul, 1984; Blankinship et al., 2014). The CO2 emitted from soil following microbial metabolic inhibition was previously ascribed to active oxidative extracellular enzymes (EXOMET) already present in soil or released from dead cells (Maire et al., 2013). However, since this study solely traced catabolism, the ultimate source of the emitted CO2 could not be definitively concluded.
This study aimed to establish the origin and underlying mechanisms of persistent CO2 release from soils exposed to model toxicants inhibiting respiratory chains. Analysis of position-specific 13C patterns in CO2, soil, microbial biomass and phospholipid-derived fatty acid (PLFA) was performed to elucidate mechanisms underlying inhibition-induced CO2 release. Measuring the production of de novo formed microbial compounds was used as criteria to confirm or reject existence of intracellular metabolism following inhibition. Production of PLFAs, components of microbial cell membranes only formed by intact and proliferating cells, is frequently utilized to confirm the presence of active intracellular metabolism. We hypothesized that a lack of 13C incorporation from labeled glucose into microbial biomass and PLFA after inhibition would confirm the proposed EXOMET theory.
Materials and methods
Sampling site
The soil was sampled from agriculturally used loamy Luvisol in northern Bavaria (49°54′ northern latitude; 11°08′ eastern longitude, 500 a.s.l.) with a mean annual temperature of 7 °C, and a mean annual precipitation of 874 mm. The soil had a pH (KCl) of 4.88, a pH (H2O) of 6.49, a TOC and TN content of 1.77% and 0.19%, respectively. Cation exchange capacity was 13 cmolC kg−1. The soil was collected from 0–10 cm, air dried, sieved to 2 mm and stored at 5 °C until use.
Experimental design
Incubations were conducted in screw-cap glass microcosms with a base layer of quartz sand. Eighty grams of dry soil was transferred to soil sample rings and installed on ceramic plates above the quartz sand. Half of the rings received NaN3 to inhibit microbial activity while the second set was not treated with NaN3 (Control). The 10 ml of 2 mm NaN3 solution added to each ring was subdivided into two portions: (1) 2 ml was added directly onto the soil surface and (2) 8 ml to the sand (to be taken up through the ceramic plates) while control soils received 10 ml of water. The added volume of water rewetted the soil to field capacity. All microcosms were preconditioned for 24 h at 5 °C. This temperature was used to eliminate interference of the CO2 determination by volatile HN3 formed from the conversion of NaN3 in soil. Four position-specific 13C-labeled isotopomeres of glucose (13C-1, 13C-2, 13C-4 and 13C-6), uniformly 13C-labeled (U-13C) and non-labeled glucose (natural abundance background) were applied to the soil in separate microcosms with four replicates each and three sampling dates resulting in 144 individual microcosms. Five milliliter of 2.55 mm glucose solution was applied onto the soil surface in each microcosm. Glucose solution for the treated soil contained 1 mm NaN3 to maintain continued inhibition. Cups filled with 5 ml of 1 m NaOH were placed into each microcosm to trap CO2. Microcosms were sealed and incubated at 5 °C in the dark.
Sampling and sample preparation
NaOH in the vials was sampled and replaced after 10 h, 1, 2, 3, 6 and 10 days. Soils were sampled after 1, 3 and 10 days. Each sample was divided into two fractions: 30 g of each sample was immediately subjected to chloroform-fumigation extraction as described below, while the remainder was stored at −20 °C for PLFA analysis.
Analytical methods
Amount and δ13C value of CO2
About 0.4 ml of each CO2 trap was diluted 1:10 with ultrapure water and CO2 content was determined by a non-dispersive infrared (NDIR) gas analyzer (TOC 5050; Shimadzu Corporation, Kyoto, Japan). The remaining volume was precipitated with 5 ml of 0.5 m SrCl2 solution. Precipitates of SrCO3 were separated by fourfold centrifuging at 2000 × g for 10 min and washing in between with millipore water to remove NaOH until pH 7 was reached. Dried SrCO3 samples (1–2 mg) were loaded into tin capsules and δ13C value was measured with a Flash 2000 Elemental analyzer coupled by a ConFlo III interface to a Delta V advantage Isotope Ratio Mass Spectrometer IRMS (all units from Thermo Fisher Scientific, Bremen, Germany). 13C respired from the applied glucose was calculated according to a mixing model (Equations (1) and (2)), where the C content of the background ([C]BG) in Equation (1) was determined by Equation (2) (Gearing, 1991)


where [C]CO2/BG/appG C content of the sample/background/applied glucose (mg C g−1soil); rCO2/BG/appG 13C atom %-excess of labeled sample/background/applied glucose (at%).
Bulk soil C and 13C content measurement
Aliquots of samples were freeze dried, ground in ball mill and 13–15 mg were weighted into tin capsules. Carbon stable isotope measurement were performed with EuroVektor elemental analyzers (HEKAtech GmbH, Wegberg, Germany) coupled by a ConFlo III interface to a Delta Plus XP IRMS (both units from Thermo Fisher Scientific). Incorporation of 13C from applied glucose into soil was calculated according to Equations (1) and (2).
Microbial biomass 13C determination
Microbial biomass C and δ13C values were determined by chloroform fumigation extraction. Soil samples were divided into two subsets of 12 g each. One subset was extracted directly and the other subset was first fumigated with chloroform for 3 days in a desiccator to lyse microbial cells. Organic C was extracted with 36 ml of 0.05 m K2SO4 on an orbital shaker for 1.5 h. Samples were centrifuged for 10 min at 2000 r.p.m. and the supernatant was filtered and frozen at −20 °C until C content analysis on a TOC/TIC analyzer (Multi C/N 2100 AnalytikJena, Jena, Germany). Thereafter, the extracts were freeze dried and about 25 mg (fumigated) and 40 mg (unfumigated) were used for δ13C values determination via EA-IRMS. Incorporation of 13C into fumigated and unfumigated samples was calculated using Equations (1) and (2). Microbial biomass C was calculated by subtracting unfumigated from fumigated C and dividing the product by a correction factor of 0.45 (Wu et al., 1990).
PLFA extraction and analysis
PLFA extraction and purification. PLFAs were analyzed according to a modified method by Frostegård et al. (1991) with each step described in detail in Gunina et al. (2014). Briefly, 25 μg of an internal recovery standard (phosphatidylcholine-dinonadecanoic acid) was added to 6 g frozen soil. PLFAs were repeatedly extracted (first with 18 ml, then with 6 ml) with a 2:1:0.8 mixture of methanol, chloroform and 0.15 m citric acid adjusted to pH 4. A two-phase mixture was generated from the combined extracts by addition of chloroform and citric acid. After shaking the lower chloroform phase was removed by liquid–liquid extraction. Neutral and glycolipids were separated from phospholipids on a solid phase extraction column packed with activated silica gel by elution with chloroform, acetone and methanol, respectively. Phospholipids were hydrolyzed in 0.5 m NaOH in methanol for 10 min on 100 °C and derivatized to fatty acid methyl esters by heating with 10% boron trifluoride in methanol at 80 °C for 15 min. Fifteen micrograms of the second external standard (tridecanoate methyl ester) together with 185 μl toluene were added to the sample and then transferred into 1.5 ml GC vials.
Analysis of δ13C values on GC-C-IRMS. One microliter of PLFA samples were injected with a 1 min splitless time into a liner at 280 °C of a Trace GC coupled via a GC-C III interface to a Delta plus IRMS (all units from Thermo Fisher Scientific). Peak separation was accomplished with two capillary columns (DB1-MS, 15 m, 250 μm ID, 0.25 μm film thickness and DB-5 MS, 30 m 250 μm ID, 0.25 μm film thickness) with helium (He 99.99% pure) as carrier gas at a flow rate of 1.7 ml per min.
Fatty acid methyl esters peaks were integrated and the δ13C values (‰) calculated via ISODAT 2.0. Drift correction was performed via repeated injection of the reference gas (CO2 99.995% pure) during measurement by linear regressions between the gas peaks surrounding the sample peaks. Correction functions according to Glaser and Amelung (2002) were used to account for the unknown δ13C value of the derivatization agents and concentration-dependent isotopic fractionation during the measurement (Schmitt et al., 2003). PLFA-C was quantified by (1) relating each fatty acid methyl esters area to the area of IS 2, (2) calculating calibration curves by linear regression of external standards consisting of 27 fatty acids at five increasing concentrations (see Supplementary Table S1) and (3) correcting for the recovery of the initially added phospholipid standard.
Statistical analysis
A Nalimov outlier test was performed for the respiration data with significance levels of 95% in case of four replicates. Microbial grouping was done by factor analysis of relative PLFA amounts (Apostel et al., 2013; Gunina et al., 2014). PLFA were grouped to one microbial group if they loaded on the same factor higher than 0.5 and if literature data on pure cultures proved their common origin (Zelles, 1999; Zelles et al., 1995). Incorporation of 13C into bulk soil, microbial biomass, PLFAs and into CO2 was tested for significant differences between the positions, incubation time and effect of NaN3 addition via factorial analysis of variance (ANOVA). If assumptions of normal distribution and homogeneous variances were not met, then outcomes were validated by a nonparametric Kruskal Wallis ANOVA. Significant differences were determined with Tukey Honest Significance Difference (Tukey HSD) post hoc test at a confidence level of 99.95%. Statistical tests were performed with Statistica (version 12.0; Statsoft GmbH, Hamburg, Germany).
Results
CO2 efflux and 13C recovery
Ten days after glucose application, total CO2 respired from azide-treated soil was twofold lower compared with control (Figure 1, top left). Two stages of glucose mineralization independent of inhibition were observed: (1) high 13C recovery in CO2 within the first 3 days and (2) low 13C recoveries thereafter (Figure 1, bottom). There were clear differences in 13C recovery from the individual glucose positions in CO2, especially in the first phase. Position-specific 13C recovery in CO2 followed a classical pattern characteristic of high pentose phosphate pathway activity, with C-1>C-4>C-2>C-6, irrespective of inhibition. On average, 13C recovery in CO2 was 12% higher in inhibited soil compared with control (Figure 1, bottom), with the largest difference occurring within the first day (Figure 1, top right).

Cumulative CO2 (mean±s.e.) from 13C uniformly labeled glucose respired during 10 days in control and NaN3-treated soils (top left), +NaN3:Control 13C (mean±s.e.) respiration rate ratio from individual glucose position (top right) and cumulative 13C (mean±s.e.) recovered in CO2 released from position-specific 13C-labeled glucose applied to soil with control (bottom left) and azide-exposed (bottom right). 13C curves were fitted with nonlinear least-square regressions according to an exponential equation (cum13C(t)=13Cmax*(1−e−kt)), where cum 13C (t) is the cumulative 13C amount depending on time, 13Cmax is the parametrically determined maximum of 13C, k is the mineralization rate and t is time (parameter estimates in Supplementary Table S4). Steven’ runs test for the fitted 13C curves revealed no deviation from linearity (Supplementary Table S5). Significant differences between fitted curves are displayed in Supplementary Table S6.
Glucose 13C incorporation into bulk soil and microbial biomass
Azide addition resulted in alteration of the 13C recovery patterns. At day 1, 13C recoveries from C-2 and C-4 in inhibited bulk soil were 17% and 20% lower compared with control (Figure 2). 13C recovery in bulk soil with inhibition did not differ between day 1 and 3 but decreased by over 12% at day 10 for each position, suggesting a shift in C transformations between day 3 and 10. In contrast, 13C recovery in control soil did not differ between the days for each glucose position. Moreover, the 13C recovery in bulk soil was five times higher than in microbial biomass, 3 days after inhibition (Figure 2), whereas in control only twice as much 13C was in bulk soil than in microbial biomass after 3 days.

13C recovery (mean±s.e.) from various glucose positions in bulk soil and extractable microbial biomass, 1, 3 and 10 days after application in control (left) and azide-exposed soils (right). Significant effects of NaN3 addition, days and individual glucose positions, according to Tukey Honest Significance Difference (Tukey HSD) post-hoc test, in bulk soil (BS) are indicated by upper case letters above the error bars, while extractable microbial biomass (MB) by lower case letters.
Position-specific patterns of 13C incorporation into microbial biomass were similar irrespective of respiration inhibition by NaN3. However, 13C recovery was threefold lower in microbial biomass after inhibition compared with control (Figure 2).
Microbial community composition
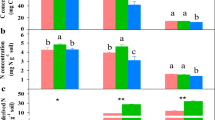
Grouping of PLFAs resulted in 10 functional microbial groups (Supplementary Table S2). In general, the biomarkers of Gram positive 2 (G+2) were dominant followed by those of Gram negative 2 (G-2) irrespective of inhibition (Figure 3, left). At day 1, the amounts of fatty acids extracted were over twofold lower in inhibited soils compared with control (Figure 3, left). Ten days after inhibition, fatty acid contents doubled to levels similar to control and pattern of fatty acid did not differ between treatments suggesting complete recovery of the microbial community in inhibited soil.

Absolute PLFA contents (mean±s.e.) of microbial groups (μg g−1 dry soil), grouped according to a factor analysis (factor loading see Supplementary Table S2) in control (open markers) and azide-exposed (solid markers) (left) and fungal/bacterial ratios (mean±s.e.) (right). The letters indicate significant differences (P<0.05) between incubation time (within microbial groups’ fatty acid content) and the effect of NaN3 addition. Meaning of microbial group acronyms are: G-1=Gram negative 1, G-2=Gram negative 2, G+1=Gram positive 1, G+2=Gram positive 2, G+3=Gram positive 3, Ac1=Actinomycets 1, Ac2=Actinomycetes 2, 18:1w9=fatty acid not associated with any microbial group, SF=saprophytic fungi and AMF=arbuscular micorrhizal fungi.
The fungal/bacterial (Fu/Ba) ratio did not differ between the days in control soils (Figure 3, right). In azide-treated soil, however, Fu/Ba ratio increased twofold between day 1 and 3, attaining similar levels at day 3 in both treatments and remained constant till day 10 (Figure 3, right).
Glucose 13C uptake by microbial groups
In control soil, 13C recovery was highest in PLFAs of G-2 and G+2, with each group containing approximately 0.09% of the applied glucose 13C in their membrane fatty acids. The other groups only incorporated between 0.01 and 0.04% of applied 13C. Incorporation of 13C into the PLFA of each microbial group did not differ between the days in both treatments (Figure 4).

13C recovery (mean±s.e.) from applied uniformly labeled 13C glucose in PLFAs of microbial groups in control (open markers) and azide-exposed (solid markers). Meaning of microbial group acronyms are: G-1=Gram negative 1, G-2=Gram negative 2, G+1=Gram positive 1, G+2=Gram positive 2, G+3=Gram positive 3, Ac1=Actinomycets 1, Ac2=Actinomycetes 2, 18:1w9=fatty acid not associated with any microbial group, SF=saprophytic fungi and AMF=arbuscular micorrhizal fungi.
In respiration-inhibited soil, however, there was no 13C incorporation into microbial PLFAs within the first 3 days (Figure 4). After 10 days, less than 0.007% of applied 13C was recovered in PLFAs of G+2, Actinomycetes 1 and 2 (Ac1 and Ac2; Figure 4). The other microbial groups did not incorporate measurable 13C into PLFAs 10 days after azide addition despite an increase in their fatty acid content (Figure 3, left), revealing dependence on other C sources for their growth.
Discussion
Total CO2 efflux and glucose mineralization
CO2 is persistently released from soil exposed to respiration-inhibiting toxicants (for example, NaN3) or heavy metals inhibiting respiration (for example, cadmium (Cd), mercury (Hg) and copper (Cu); Ausmus et al., 1978; Rozycki and Bartha, 1981; Fliessbach et al., 1994; Trevors, 1996). Application of 13C glucose to NaN3-inhibited and control soils resulted in high 13C recovery in CO2 within the first 3 days (Figure 1, bottom), consistent with intense glucose mineralization. Lower 13C recoveries thereafter reflect mineralization of glucose-derived metabolites after exhaustion of glucose (Blagodatskaya et al., 2011). The 13C recovery pattern in CO2 was similar (C-1>C-4>C-2>C-6) under both conditions. The high 13C recovery from glucose C-1 position in CO2 reveals that glucose was predominantly catabolized via the pentose phosphate pathway (Caspi et al., 2008; Dijkstra et al., 2011b; Apostel et al., 2015).
Average 13C recovery in CO2 was 12% higher from inhibited soil compared with control. In contrast, total CO2 from inhibited soil was half emitted from control (Figure 1, top left). High CO2 release with low 13C content from control points towards a priming effect, that is, a glucose-induced acceleration of SOM decomposition (Kuzyakov, 2010; Blagodatskaya and Kuzyakov, 2013). Increased 13C recovery and reduced total CO2 after NaN3 inhibition could not be explained by a shift towards fermentative metabolism resulting from respiration inhibition, as 13CO2 from fermentation would be C-4 dominated instead of C-1. High CO2 emission from soils with minimized microbial activity can be attributed to release of active oxidative extracellular enzymes (EXOMET) from dead organisms (Maire et al., 2013). To prove or reject the relevance of EXOMET compared with cellular metabolism, investigation of intracellularly formed metabolites (that is, de novo formed microbial biomass) was conducted.
13C incorporation into microbial biomass and bulk soil
Glucose is utilized by microorganisms for biosynthesis of various cellular building blocks and as an energy source (Gunina et al., 2014). Recovery of glucose-derived 13C in microbial biomass is indicative of active intracellular biosynthetic processes. 13C recovery in microbial biomass from C-6, C-4 and C-2 was higher than C-1 in control soil (Figure 2, left), with a similar pattern arising in control bulk soil, confirming that microbial products, and not untransformed glucose, are the predominant source of extracellular 13C. The position-specific 13C recovery pattern in bulk soil and microbial biomass complemented the metabolic fluxes observed in CO2 and confirm that glucose was predominantly metabolized via the pentose phosphate pathway. The dominance of this pathway at 5 °C under moderate C supply reflects the classical metabolic C allocation observed in previous studies (Dijkstra et al., 2011b). Temperature decrease shifts metabolic activity from glycolysis and NADH production to the pentose phosphate pathway and NADPH production in microbial cultures (Wittmann et al., 2007) to meet the pentose and NADPH demands for biosynthesis (Fuhrer and Sauer, 2009). High 13C recovery from positions C-6, C-4 and C-2 implies that, after loss of the C-1, the remaining part of the molecule was allocated to biosynthesis (Gunina et al., 2014). Levels of 13C in microbial biomass did not change between the days in control soils, suggesting that glucose was consumed within the first day and was incorporated into C pools with slower turnover.
In inhibited soil, glucose 13C recovery from C-6 and C-2 in bulk soil and microbial biomass was higher than of C-1 and C-4 (Figure 2, right), suggesting intracellular transformation to pyruvate via the pentose phosphate pathway (Dijkstra et al., 2015) followed by oxidization for energy production. Increased glucose oxidation after pyruvate formation also accounts for the altered pattern of 13C recovery observed in bulk soil, with even stronger decrease in C-4 and C-2 incorporation at day 1 compared with control soil (Dijkstra et al., 2015). Intracellular transformation of glucose after NaN3 inhibition opposes the concept of extracellular oxidative metabolism (Maire et al., 2013), as surviving microbes must be responsible for the observed transformation. The ≈3-fold lower 13C recovery in microbial biomass and higher recovery in CO2 following inhibition compared with control conditions (Figure 1 and 2, bottom) imply that the surviving microbes utilized substantial amount of glucose for energy production. This shift towards energy production may represent an adaptation mechanism by tolerant bacterial strains to overcome inhibition, as microorganisms that survive heavy metal intoxication also divert a large amount of energy from added substrate towards energy-intensive physiological detoxification mechanisms (Gordon et al., 1993). Resistance to toxicants shifts microbial community structure to compensate for the loss of more sensitive populations (Giller et al., 1998). Furthermore, resistance of some Trichoderma strains to NaN3 was reported by Kelley and Rodriguez-Kabana (1981). Therefore, we conducted PLFA analysis to identify those microbial groups that survive inhibition.
13C incorporation into PLFAs
13C was incorporated into PLFAs of each microbial group in control soils with G-2 and G+2 incorporating two times more than other microbial groups (Figure 4). At room temperatures, growth rate of Gram-negative bacteria is dependent upon the concentration of easily available substrate (Treonis et al., 2004). Therefore, they dominate the rhizosphere, where low molecular weight organic substances are present in high concentrations (Gunina et al., 2014; Apostel et al., 2015). In contrast, Gram-positive bacteria are typically abundant in bulk soil and incorporate C from old SOM (Kramer and Gleixner, 2006). Therefore, 13C recovery within the same range in PLFA of G-2 and G+2 in control soils implies that Gram-positive bacteria profited from lower competitiveness of Gram negatives at low temperature (5 °C). The lack of difference in 13C incorporation into PLFAs between the days was similar to results in microbial biomass, confirming complete glucose incorporation within the first day and slow transformation of 13C in microbial products thereafter.
Ten days after inhibition, incorporation of glucose-derived 13C into fatty acids was only observed in G+2 and actinomycetes (Figure 4), that is, groups known to use old SOM or microbial necromass (McCarthy and Williams, 1992; Kramer and Gleixner, 2006). Therefore, their 13C incorporated into PLFA is likely to occur by recycling of glucose-derived excretion products and not intact glucose. Such recycling of extracellular glucose-derived metabolites explains the decrease in 13C recovery by over 12% in bulk soil 10 days after inhibition. There was no detectable glucose 13C incorporation in any PLFA 3 days after inhibition (Figure 4), despite more than 9% recovery in microbial biomass, suggesting either (1) absence of growth and membrane repair or (2) glucose C was not allocated to fatty acid biosynthesis. To determine the most likely scenario, we examined the fatty acid content of microbial biomass to determine the growth status of the microbial populations.
Effects of NaN3 on microbial community structure
Comparing the fungal/bacterial ratio in inhibited and control soils at day 1 demonstrated a greater short-term susceptibility of fungi to azide inhibition (Figure 3, right). However, a twofold increase of the fungi/bacteria ratio to a level equal to control soils within 3 days shows fast recovery of fungal biomass.
The fatty acid contents associated with each microbial group were more than twofold lower after inhibition compared with control soil at day 1, likely resulting from cell asphyxiation (Winter et al., 2012). However, the twofold increase in fatty acid content associated with each microbial group in inhibited soils between day 1 and 10 implies that there was growth of the affected microbial groups. This contradicts previous assumptions that microorganisms stressed by exposure to toxicants divert energy from growth to maintenance (Killham, 1985). Furthermore, the increasing fatty acid contents raise the question how microorganisms grow without incorporating glucose into their PLFA. Living microorganisms can utilize microbial necromass from soil (Dippold and Kuzyakov, 2016; Figure 5), whose pool was likely substantial resulting from the death of a high percentage of microorganisms after inhibition. The energy for transforming or recycling of such compounds was provided by an increase in glucose oxidation, which was proven by the 12% increased 13C recovery in CO2 and the ≈3-fold lower 13C recovery in microbial biomass after inhibition compared with control. Preferential recycling of the cost-intensive alkyl chains of PLFA and other cost-intensive biomass compounds occur under such conditions. This recycling is accompanied by de-novo formation of other cheaper biomass compounds. Therefore, ecosystems are self-regulating systems that evolve mechanisms of self-repair and their biological populations are adapted to resist and recover from environmental fluctuations (EFSA, 2016). Diminishing effects of inhibition over time leads to potential recovery of the microbial groups (EFSA, 2016) as indicated by increase in PLFA content.

Microbial glucose transformation pathways in control (left) and mechanisms adapted to overcome inhibition (right). Colored arrows correspond to glucose C positions and indicate their fate. The thickness of the colored arrows towards CO2 is proportional to amount. Black straight and broken arrows indicate metabolites formed or recycled. Black curvy arrows indicate redox processes.
Adaptation mechanisms to respiration inhibition
Azide inhibits electron transfer in non-phosphorylating submitochondrial particles at cytochrome oxidase and catalase (Keilin, 1936) resulting in cell asphyxiation (Winter et al., 2012). Continued respiration after inhibition, with intensive intracellular transformation via the pentose phosphate pathway raises the question: How do microorganisms manage intracellular respiration without electron acceptors (NAD+ or NADP+), which cannot be regenerated after azide inhibition? A common response of bacteria to electron–acceptor limitation is to produce electrically conductive pilus-like appendages called bacterial nanowires (Reguera et al., 2005; Gorby et al., 2006), which anchor between the periplasmic and outer membranes and allow transfer of electrons from the cell to minerals containing Fe3+ and Mn4+ in the extracellular environment (Reguera et al., 2005; Gorby et al., 2006; Figure 5). Other demonstrated mechanisms to overcome inhibition include (1) electron shuttling between the cell and extracellular minerals via humic substances in solution (Lovley et al., 1996; Bi et al., 2013; Piepenbrock et al., 2014) and solid state (Roden et al., 2010) or (2) excretion of reduced metabolites by microorganisms, including quinones (Newman and Kolter, 2000) and phenolic compounds (Vempati et al., 1995; Pentrakova et al., 2013), that transfer electrons to the extracellular environment (Figure 5). Processes outlined above demonstrate that electron transport chain inhibition does not stop intracellular microbial metabolism in soil. Without compound-specific 13C measurement of the extracellular metabolite pool, it is not possible to point out which of these processes for extracellular electron disposal dominated after NaN3 addition. However, extracellular electron transfer by excretion of reduced metabolites explains the high 13C recovery in the bulk soil compared with microbial biomass during the first 3 days following inhibition. The effectiveness of NaN3 as a bacteriostat in liquid samples such as antibodies, milk (Winter et al., 2012) or water samples does not contradict our findings, because such samples lack humic substances or minerals (Fe3+ and Mn4+) permitting extracellular electron transfer via the above-mentioned mechanisms.
Previously, persistent CO2 release from soil with eliminated or inhibited respiration was attributed to abiotic processes (Rozycki and Bartha, 1981; Trevors, 1996) and extracellular oxidative metabolism (Maire et al., 2013). Our data clearly demonstrate that inhibition of electron transport chain cannot stop intracellular glucose metabolism: microorganisms circumvent respiration inhibition via temporary extracellular electron transfer, giving organisms a chance to reconstruct new electron transport chains and resume normal (aerobic) respiration. Thus, even under respiration inhibition no indication for extracellular glucose metabolism could be perceived. The ability of microorganisms to overcome limitations of intracellular metabolism by utilizing SOM resources is important for understanding the origin of CO2 emitted from soils with respiration-inhibiting toxicants (Bond et al., 1976; Ausmus et al., 1978; Trevors, 1996). Heavy metals such as Cd, Hg and Cu, which are widespread in the environment due to their industrial, agricultural, medical and technological application, inhibit respiration (Belyaeva et al., 2012). However, their presence in organic or clayey soils does not stop respiration (Bond et al., 1976; Ausmus et al., 1978; Fliessbach et al., 1994), a phenomenon that could similarly be explained by extracellular electron transfer because such soils are rich in humic substances and minerals (Fe3+ and Mn4+) functioning as extracellular electron acceptors. Microorganisms’ tolerance and resistance to copper-based fungicides could also be attributed to extracellular electron transfer. Therefore, the mechanisms proven from metabolic perspective in this study contribute to a better understanding of microbial resilience and resistance to direct respiration-inhibiting toxicants (for example, azides, cyanides), pesticides (for example, phosphides and phosphines) or heavy metals (for example, Cu and Hg) and subsequent ecological recovery after perturbation. Such mechanisms are limited to portion of microorganisms activated by glucose (Monard et al., 2008) and toxicants that inhibit respiratory chains directly.
Conclusions
Combining position-specific 13C labeling with compound-specific 13C-PLFA analysis proved to be a valuable tool to understand how microorganisms overcome respiration inhibition. Glucose was metabolized by soil microorganisms via the pentose phosphate pathway irrespective of respiration inhibition. NaN3 reduced total CO2 efflux twofold but increased 13C recovery in released CO2 by 12% compared with control soils. The low 13C recovery in microbial biomass increased pyruvate oxidation and increased proportion of glucose-derived metabolites in extracellular microbial products following NaN3 application provide evidence for increased glucose use for energy production and synthesis of extracellular electron transport compounds to bypass inhibition. Resources for growth were recycled from the large pool of microbial necromass resulting from toxicant addition. Consequently, to overcome intracellular inhibition of the electron transport chain, microorganisms most likely coupled intracellular metabolism with extracellular redox processes. This is possible only in soil and similar environments rich in electron acceptors. Construction of new electron transport chains and resumption of aerobic respiration as well as recovery of microbial groups occurred within 10 days at 5 °C. We assume this bypass of respiration inhibition will be much faster at high temperatures.
These results suggest that the persistent CO2 efflux after azide addition to soil is as a result of intracellular oxidation of SOM followed by extracellular electron disposal. This mechanism is also likely to account for microbial tolerance to, for example, heavy metals and other toxicants directly altering microbial respiration in soils but requires confirmation by extending this position-specific labeling approach on soils contaminated with a broad spectrum of toxicants. Finally, this metabolic tracing approach provides an understanding of the impacts of chemicals such as azides, cyanides and heavy metals on soil C cycling following contamination and enables development of unique insights concerning soil-specific microbial mechanisms to overcome respiration inhibition.
References
Apostel C, Dippold M, Glaser B, Kuzyakov Y . (2013). Biochemical pathways of amino acids in soil: assessment by position-specific labeling and 13C-PLFA analysis. Soil Biol Biochem 67: 31–40.
Apostel C, Dippold M, Kuzyakov Y . (2015). Biochemistry of hexose and pentose transformations in soil analyzed by position-specific labeling and 13C-PLFA. Soil Biol Biochem 80: 199–208.
Ausmus BS, Dodson GJ, Jackson DR . (1978). Behavior of heavy-metal in forest microcosms. 3. Effects on litter-carbon metabolism. Water Air Soil Pollut 10: 19–26.
Babich H, Stotzky G . (1985). Heavy metal toxicity to microbe-mediated ecologic processes: a review and potential application to regulatory policies. Environ Res 36: 111–137.
Belyaeva EA, Sokolova TV, Emelyanova LV, Zakharova IO . (2012). Mitochondrial electron transport chain in heavy metal-induced neurotoxicity: effects of cadmium, mercury, and copper. Sci World J 2012: 136063–136063.
Bi R, Lu Q, Yu W, Yuan Y, Zhou S . (2013). Electron transfer capacity of soil dissolved organic matter and its potential impact on soil respiration. J Soil Sediment 13: 1553–1560.
Blagodatskaya E, Kuzyakov Y . (2013). Active microorganisms in soil: critical review of estimation criteria and approaches. Soil Biol Biochem 67: 192–211.
Blagodatskaya E, Yuyukina T, Blagodatsky S, Kuzyakov Y . (2011). Three-source-partitioning of microbial biomass and of CO2 efflux from soil to evaluate mechanisms of priming effects. Soil Biol Biochem 43: 778–786.
Blankinship JC, Becerra CA, Schaeffer SM, Schimel JP . (2014). Separating cellular metabolism from exoenzyme activity in soil organic matter decomposition. Soil Biol Biochem 71: 68–75.
Bond H, Lighthart B, Shimabuku R, Russell L . (1976). Some effects of cadmium on coniferous forest soil and litter microcosms. Soil Sci 121: 278–287.
Bremer E, van Kessel C . (1990). Extractability of microbial 14C and 15N following addition of variable rates of labelled glucose and (NH4 2SO4 to soil. Soil Biol Biochem 22: 707–713.
Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M et al. (2008). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res 36: D623–D631.
Dijkstra P, Dalder JJ, Selmants PC, Hart SC, Koch GW, Schwartz E et al. (2011a). Modeling soil metabolic processes using isotopologue pairs of position-specific C-13-labeled glucose and pyruvate. Soil Biol Biochem 43: 1848–1857.
Dijkstra P, Salpas E, Fairbanks D, Miller EB, Hagerty SB, van Groenigen KJ et al. (2015). High carbon use efficiency in soil microbial communities is related to balanced growth, not storage compound synthesis. Soil Biol Biochem 89: 35–43.
Dijkstra P, Thomas SC, Heinrich PL, Koch GW, Schwartz E, Hungate BA . (2011b). Effect of temperature on metabolic activity of intact microbial communities: evidence for altered metabolic pathway activity but not for increased maintenance respiration and reduced carbon use efficiency. Soil Biol Biochem 43: 2023–2031.
Dippold M, Biryukov M, Kuzyakov Y . (2014). Sorption affects amino acid pathways in soil: implications from position-specific labeling of alanine. Soil Biol Biochem 72: 180–192.
Dippold MA, Kuzyakov Y . (2013). Biogeochemical transformations of amino acids in soil assessed by position-specific labelling. Plant Soil 373: 385–401.
Dippold MA, Kuzyakov Y . (2016). Direct incorporation of fatty acids into microbial phospholipids in soils: position-specific labeling tells the story. Geochim Cosmochim Acta 174: 211–221.
EFSA. (2016). Recovery in environmental risk assessments at EFSA. EFSA J 14: 4313.
Fischer H, Ingwersen J, Kuzyakov Y . (2010). Microbial uptake of low-molecular-weight organic substances out-competes sorption in soil. Eur J Soil Sci 61: 504–513.
Fischer H, Meyer A, Fischer K, Kuzyakov Y . (2007). Carbohydrate and amino acid composition of dissolved organic matter leached from soil. Soil Biol Biochem 39: 2926–2935.
Fliessbach A, Martens R, Reber HH . (1994). Soil microbial biomass and microbial activity in soils treated with heavy metal contaminated sewage sludge. Soil Biol Biochem 26: 1201–1205.
Frostegård Å, Tunlid A, Bååth E . (1991). Microbial biomass measured as total lipid phosphate in soils of different organic content. J Microbiol Methods 14: 151–163.
Fuhrer T, Sauer U . (2009). Different biochemical mechanisms ensure network-wide balancing of reducing equivalents in microbial metabolism. J Bacteriol 191: 2112–2121.
Gearing JN . (1991). The study of diet and trophic relationships through natural abundance 13C. In: Coleman DC, Fry B (eds). Carbon Isotope Techniques. Academic Press: New York, pp 201–218..
Giller KE, Witter E, McGrath SP . (1998). Toxicity of heavy metals to microorganisms and microbial processes in agricultural soils: a review. Soil Biol Biochem 30: 1389–1414.
Glaser B, Amelung W . (2002). Determination of C-13 natural abundance of amino acid enantiomers in soil: methodological considerations and first results. Rapid Commun Mass Spectrom 16: 891–898.
Gorby YA, Yanina S, McLean JS, Rosso KM, Moyles D, Dohnalkova A et al. (2006). Electrically conductive bacterial nanowires produced by Shewanella oneidensis strain MR-1 and other microorganisms. Proc Natl Acad Sci USA 103: 11358–11363.
Gordon AS, Harwood VJ, Sayyar S . (1993). Growth, copper-tolerant cells, and extracellular protein production in copper-stressed chemostat cultures of Vibrio alginolyticus. Appl Environ Microbiol 59: 60–66.
Gunina A, Dippold MA, Glaser B, Kuzyakov Y . (2014). Fate of low molecular weight organic substances in an arable soil: from microbial uptake to utilisation and stabilisation. Soil Biol Biochem 77: 304–313.
Gunina A, Kuzyakov Y . (2015). Sugars in soil and sweets for microorganisms: review of origin, content, composition and fate. Soil Biol Biochem 90: 87–100.
ISO 16072. (2002) Soil quality—Laboratory Methods for Determination of Microbial Soil Respiration. International Organization for Standardization: Geneva, Switzerland.
Keilin D . (1936). The action of sodium azide on cellular respiration and on some catalytic oxidation reactions. Proc Roy Soc Lond B Biol Sci 121: 165–173.
Kelley WD, Rodriguez-Kabana R . (1981). Effects of annual applications of sodium azide on soil fungal populations with emphasis on Trichoderma species. Pestic Sci 12: 235–244.
Killham K . (1985). A physiological determination of the impact of environmental stress on microbial bomass. Environ Pollut A 38: 283–294.
Kramer C, Gleixner G . (2006). Variable use of plant- and soil-derived carbon by microorganisms in agricultural soils. Soil Biol Biochem 38: 3267–3278.
Kuzyakov Y . (2010). Priming effects: interactions between living and dead organic matter. Soil Biol Biochem 42: 1363–1371.
Lovley DR, Coates JD, BluntHarris EL, Phillips EJP, Woodward JC . (1996). Humic substances as electron acceptors for microbial respiration. Nature 382: 445–448.
Maire V, Alvarez G, Colombet J, Comby A, Despinasse R, Dubreucq E et al. (2013). An unknown oxidative metabolism substantially contributes to soil CO2 emissions. Biogeosciences 10: 1155–1167.
Mau RL, Liu CM, Aziz M, Schwartz E, Dijkstra P, Marks JC et al. (2015). Linking soil bacterial biodiversity and soil carbon stability. ISME J 9: 1477–1480.
McCarthy AJ, Williams ST . (1992). Actinomycetes as agents of biodegradation in the environment—a review. Gene 115: 189–192.
Monard C, Binet F, Vandenkoornhuyse P . (2008). Short-term response of soil bacteria to carbon enrichment in different soil microsites. Appl Environ Microbiol 74: 5589–5592.
Newman DK, Kolter R . (2000). A role for excreted quinones in extracellular electron transfer. Nature 405: 94–97.
Pentrakova L, Su K, Pentrak M, Stuck JW . (2013). A review of microbial redox interactions with structural Fe in clay minerals. Clay Miner 48: 543–560.
Piepenbrock A, Schroeder C, Kappler A . (2014). Electron transfer from humic substances to biogenic and abiogenic Fe(III) oxyhydroxide minerals. Environ Sci Technol 48: 1656–1664.
Reguera G, McCarthy KD, Mehta T, Nicoll JS, Tuominen MT, Lovley DR . (2005). Extracellular electron transfer via microbial nanowires. Nature 435: 1098–1101.
Roden EE, Kappler A, Bauer I, Jiang J, Paul A, Stoesser R et al. (2010). Extracellular electron transfer through microbial reduction of solid-phase humic substances. Nat Geosci 3: 417–421.
Rozycki M, Bartha R . (1981). Problems associated with the use of azide as an inhibitor of microbial activity in soil. Appl Environ Microbiol 41: 833–836.
Scandellari F, Hobbie EA, Ouimette AP, Stucker VK . (2009). Tracing metabolic pathways of lipid biosynthesis in ectomycorrhizal fungi from position-specific 13C-labelling in glucose. Environ Microbiol 11: 3087–3095.
Schimel J . (2013). Soil carbon microbes and global carbon. Nat Climate Change 3: 867–868.
Schmitt J, Glaser B, Zech W . (2003). Amount-dependent isotopic fractionation during compound-specific isotope analysis. Rapid Commun Mass Spectrom 17: 970–977.
Treonis AM, Ostle NJ, Stott AW, Primrose R, Grayston SJ, Ineson P . (2004). Identification of groups of metabolically-active rhizosphere microorganisms by stable isotope probing of PLFAs. Soil Biol Biochem 36: 533–537.
Trevors JT . (1996). Sterilization and inhibition of microbial activity in soil. J Microbiol Methods 26: 53–59.
Vempati RK, Kollipara KP, Stucki JW, Wilkinson H . (1995). Reduction of structural iron in selected iron-bearing minerals by soybean root exudates grown in an in-vitro geoponicsystem. J Plant Nutr 18: 343–353.
Voroney RP, Paul EA . (1984). Determination of KC and KN insitu for calibration of the chloroform fumigation incubation method. Soil Biol Biochem 16: 9–14.
Winter C, Kerros M-E, Weinbauer MG . (2012). Effects of sodium azide on the abundance of prokaryotes and viruses in marine samples. PLoS One 7: e37597.
Wittmann C, Weber J, Betiku E, Kroemer J, Boehm D, Rinas U . (2007). Response of fluxome and metabolome to temperature-induced recombinant protein synthesis in Escherichia coli. J Biotechnol 132: 375–384.
Wu J, Joergensen RG, Pommerening B, Chaussod R, Brookes PC . (1990). Measurement of soil microbial biomass C by fumigation extraction—an automated procedure. Soil Biol Biochem 22: 1167–1169.
Zelles L . (1999). Fatty acid patterns of phospholipids and lipopolysaccharides in the characterisation of microbial communities in soil: a review. Biol Fertil Soils 29: 111–129.
Zelles L, Bai QY, Rackwitz R, Chadwick D, Beese F . (1995). Determination of phospholipid-derived and lipopolysaccharide-derived fatty-acids as an estimate of microbial biomass and community structures in soils. Biol Fertil Soils 19: 115–123.
Acknowledgements
We thank the DFG for funding (DI-2136/1-1 and NTS 186/1006-1/P) and DAAD for funding Ezekiel Bore. We thank the technical staff of Goettingen University, in particular Karin Schmidt and Anita Kriegel, for microbial biomass C content determination, the entire team at KOSI (Centre for Stable Isotopes Analysis) for δ13C analysis and Joshua Bostic for English proofreading.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
K Bore, E., Apostel, C., Halicki, S. et al. Soil microorganisms can overcome respiration inhibition by coupling intra- and extracellular metabolism: 13C metabolic tracing reveals the mechanisms. ISME J 11, 1423–1433 (2017). https://doi.org/10.1038/ismej.2017.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.3
This article is cited by
-
Carbendazim Modulates the Metabolically Active Bacterial Populations in Soil and Rhizosphere
Current Microbiology (2023)
-
The presence of soybean, but not soybean cropping frequency has influence on SOM priming in crop rotation systems
Plant and Soil (2023)
-
Drought re-routes soil microbial carbon metabolism towards emission of volatile metabolites in an artificial tropical rainforest
Nature Microbiology (2023)
-
Heavy metal pollution increases soil microbial carbon limitation: Evidence from ecological enzyme stoichiometry
Soil Ecology Letters (2021)
-
Biogeochemical transformation of greenhouse gas emissions from terrestrial to atmospheric environment and potential feedback to climate forcing
Environmental Science and Pollution Research (2020)
