
Abstract
The aim of this study was to extend the results of our previous stable isotope probing (SIP) investigation: we identified a soil fungus involved in phenol biodegradation at an agricultural field site. DNA extracts from our previous study were examined using fungi-specific PCR amplification of the 18S–28S internal transcribed spacer (ITS) region. We prepared an 80-member clone library using PCR-amplified, 13C-labeled DNA derived from field soil that received 12 daily doses of 13C-phenol. Restriction-fragment-length-polymorphism screening and DNA sequencing revealed a dominant clone (41% of the clone library), the ITS sequence of which corresponded to that of the fungal genus Trichosporon. We successfully grew and isolated a white, filamentous fungus from site soil samples after plating soil dilutions on mineral salts agar containing 250 p.p.m. phenol. Restreaking on both yeast extract–peptone–galactose and Sabouraud dextrose agar plates led to further purification of the fungus, the morphological characteristics of which matched those of the genus Trichosporon. The ITS sequence of our isolated fungus was identical to that of a clone from our SIP-based library, confirming it to be Trichosporon multisporum. High-performance liquid chromatography and turbidometeric analyses showed that the culture was able to metabolize and grow on 200 p.p.m. phenol in an aqueous mineral salts medium within 24 h at room temperature. Gas chromatography–mass spectrometry analysis of 13CO2 respiration from laboratory soil incubations demonstrated accelerated phenol mineralization in treatments inoculated with T. multisporum. These findings show that T. multisporum actively degraded phenol in our field-based, soil experiments.
Similar content being viewed by others

Introduction
Phenol is a globally distributed, organic pollutant that enters the environment through both natural (decomposition of organic matter, burning wood and volatilization from manure) and anthropogenic sources (coal tar waste, production of resins, plastics, adhesives, nylon, steel, rubber and treated lumber) (Scow et al., 1981; Goerlitz et al., 1985; Jenkins, 1994; ATSDR, 2006). The universal toxicity and ubiquitous nature of phenol make its biodegradation a topic of great interest (ATSDR, 2006). Although the earliest studies of phenol biodegradation focused on bacteria, there have been a growing number of fungi studied for their ability to degrade phenol, including members of the genera Trichosporon, Cryptococcus, Rhodotorula, Rhodococcus, Aspergillus, Penicillium, Cladosporium, Trichoderma, Phanerochaete, Candida and Fusarium (Harris and Ricketts, 1962; Dagley, 1967; Neujahr and Varga, 1970; Gaal and Neujahr, 1979; Rubin and Schmidt, 1985; Alexievaa et al., 2004; Atagana, 2004; Santos and Linardi, 2004; Bergauer et al., 2005; Margesin et al., 2005; Krallish et al., 2006; Singh, 2006; Stoilova et al., 2006; Jiang et al., 2007). Fungi are often able to thrive under environmentally stressed conditions (low nutrient availability, low moisture, low pH and low temperature) and unlike bacteria, they can extend their biomass through environmental matrices through hyphal growth, making their potential for metabolism of organic pollutants particularly promising (Buchan et al., 2003; Atagana, 2004; Margesin et al., 2005; de Boer et al., 2005). Singh (2006) provides a comprehensive review of phenol metabolism by fungi.
Stable isotope probing (SIP) has proven to be a valuable tool in identifying microorganisms actively degrading chemical pollutants in complex microbial communities (Jeon et al., 2003; Kasai et al., 2006; Leigh et al., 2007; Jones et al., 2008). SIP follows incorporation of a stable isotope (for example, 13C, 15N and 18O) into cellular biomarkers (phospholipid fatty acids, DNA, RNA and protein) of organisms actively involved in the metabolism of a labeled substrate (Radajewski et al., 2003; Neufeld et al., 2007b, 2007c; Schwartz, 2007; Whiteley et al., 2007; Jehmlich et al., 2008). A number of reviews are available that discuss the various applications and methodological considerations of SIP studies (Radajewski et al., 2003; Dumont and Murrell, 2005; Madsen, 2006; Neufeld et al., 2007c; Whiteley et al., 2007).
Our recent work focused on members of the domain, Bacteria, involved in phenol degradation at an agricultural field site (DeRito et al., 2005). Our methods allowed us to identify a number of primary degraders of phenol as well as potential secondary consumers involved in carbon flow between members of the soil microbial community. The current study extends our inquiry to the fungal component of soil microorganisms. Using field-based DNA SIP of the 18S–28S internal transcribed spacer (ITS) region from previous centrifugation gradients (DeRito et al., 2005), we identified a soil yeast actively involved in phenol biodegradation. Isolation of the phenol-degrading yeast, Trichosporon multisporum strain CD1, complemented our culture-independent findings and confirmed that this soil-borne fungus actively degraded phenol in our field experiments.
Materials and methods
Field study site
This study was conducted at the Cornell University Agricultural Experiment Station, Ithaca, NY, USA. The soil plot (Collamer silt loam) was level and free of vegetation. The treatments (see below) occurred in a grid of dosing points on ∼10-cm centers. A table was placed over the plot (0.8 m high) to protect the experiment from rain and direct exposure to sunlight.
Prior experimental design
Five soil treatments, prepared in quadruplicate (Table 1), were designed to probe three distinct communities of phenol degraders in situ. Key variables for each treatment were carbon isotope of phenol (unlabeled [12C] or 13C) and the number of daily doses (0 or 11) prior to a final dose of phenol. For example, in Table 1, ‘13/13’ indicates that 11 prior doses of 13C-labeled phenol were delivered and the final dose was also 13C-labeled. Each 20-μl dose contained 200 μg of phenol. Immediately after the final dose of 13C-phenol, the plots were covered with septa-fitted chambers. Following 30 h of headspace monitoring of 13CO2, 0.125-g soil samples from quadruplicate samples were pooled and stored at −80 °C. Subsequently, DNA extracts were prepared (DeRito et al., 2005).
Fungi-specific PCR
Five 13C-DNA fractions obtained from our previous phenol study (DeRito et al., 2005) were PCR amplified using the primers 1406f, 5′-TGYACACACCGCCCGT-3′ (universal, 16S/18S rRNA gene), and 3126r, 5′-ATATGCTTAAGTTCAGCGGGT-3′, by methods described earlier (Sequerra et al., 1997; Fisher and Triplett, 1999; Ranjard et al., 2001; Hansgate et al., 2005). These primers target the last 120 bp of 18S rRNA gene, ITS region 1 (ITS1), 5.8S rRNA gene and ITS2, with the 3126r primer matching the 5′ end of the 28S rRNA gene. Each 50 μl PCR reaction contained 10 μl of template DNA, 400 nM of each primer and 1 × Taq PCR Master Mix (ABGene, Rochester, NY, USA).
Cloning, restriction digestion and sequencing
Molecular cloning of the resulting fungal amplicon (from the 2 × 10−2 dilution) was carried out using the TOPO Cloning Kit (TA cloning; Invitrogen, Carlsbad, CA, USA) following the manufacturer's recommended protocol. Following transformation of plasmids into host cells and blue/white screening, colonies with inserts were verified by PCR with vector-specific primers (5′-GTAACGGCCGCCAGTGTGCT-3′ and 5′-CAGTGTGATGGATATCTGCA-3′) that flanked the cloning region. Amplicons were digested with HaeIII and HhaI and restriction fragment length polymorphism (RFLP) patterns were analyzed on 3% MetaPhore agarose gels (BioWhittaker; Molecular Applications, Rockland, ME, USA) with a 100-bp ladder (Promega, Madison, WI, USA) as a marker. Clones containing unique RFLP patterns were selected for sequencing, grown overnight in 5 ml of Luria–Bertani broth with kanamycin (50 μg μl−1) and pelleted, and plasmids were purified (QiaPrep spin miniprep kit; Qiagen, Santa Clarita, CA, USA). Sequencing (Cornell University DNA Sequencing Facility) was conducted using the vector-specific M13 forward (5′-TGTAAAACGACGGCCAGT-3′) and M13 reverse (5′-AACAGCTATGACCATG-3′) primers. Raw sequence data were assembled into full-length contigs (516–629 bp) using the program SEQMAN II (DNASTAR Inc., Madison, WI, USA). After assembly, the consensus sequence was verified manually by referring to the corresponding ABI chromatograms of the sequencing reactions. A BLAST search (http://ncbi.nlm.nih.gov/BLAST) was used to identify the closest relatives for each clone, which are included in the resulting dendrogram. Sequences were aligned using the Clustal W option in the program MEGALIGN (DNASTAR Inc.) and sequence relatedness was calculated using the same program. Phylogenetic analysis was conducted using ClustalX version 1.83.
Isolation and identification of phenol-degrading fungi
For the isolation of phenol-degrading fungi, a modified version of medium from Shoda et al. (1980) was prepared with the following composition, per liter of distilled water: phenol 0.250 g, NH4Cl 5 g, KH2PO4 5 g, MgSO4·7H2O 0.1 g, CaCl2·2H2O 0.01 g, FeCl3·6H2O 0.01 g, pyridoxine HCl 0.4 g, thiamine HCl 0.2 g, riboflavin 0.2 g, calcium pantothenate 0.2 g, thioctic acid 0.2 g, ρ-aminobenzoic acid 0.2 g, vitamin B12 0.2 g, mercaptoethanesulfonic acid 0.2 g, biotin 0.08 g and folic acid 0.08 g. To inhibit bacterial growth, kanamycin and streptomycin were added to a final concentration of 100 p.p.m. Agar plates were prepared using the above medium containing 1.5% Agar Noble (Difco).
Soil (5 g) was added to 100 ml of liquid medium containing 250 p.p.m. phenol and agitated on a rotary shaker (300 r.p.m.) for 2 days at room temperature. During the incubation, phenol loss was confirmed by high-performance liquid chromatography (HPLC) analysis and optical density (OD) increased. The enrichment culture was diluted with phosphate-buffered saline and 100 μl aliquots of each dilution (10−2–10−6) were spread onto agar plates containing 250 p.p.m. phenol and incubated at room temperature. White filamentous colonies appeared after 2 days and were homogenous at each dilution. Two representative colonies were selected and purified on yeast extract–peptone–galactose (composition per liter: 10 g Bacto yeast extract, 20 g Bacto peptone and 20 g galactose) and Sabouraud dextrose agar plates. Phenol-degrading capacity of these two isolates was confirmed by restreaking on agar plates containing phenol as the carbon source. Isolates were identified morphologically, microscopically and by sequence analysis of the fungal intergenic spacer region (Kurtzman, 1988; Cullings and Vogler, 1998; Redecker et al., 1999; Buchan et al., 2003).
Metabolism of phenol by T. multisporum strain CD1
One of the purified Trichosporon isolates (see above) was inoculated into a flask containing 100 ml of liquid media and 200 p.p.m. phenol (see above). The flask was shaken at room temperature for 48 h. Next, 200 μl of this cell culture (OD∼0.1) was inoculated into 200 ml of fresh media containing 200 p.p.m. phenol. Cell growth was measured by OD at 600 nm with a spectrophotometer (Bausch and Lomb, Rochester, NY USA). Phenol degradation was measured by HPLC analysis (see below).
T. multisporum strain CD1 was also tested for its ability to degrade phenol in soil. Two different cell inocula were prepared by growing strain CD1 to an OD600 ∼0.4 in both 10% Sabouraud dextrose broth and in minimal media containing 0.4% glucose, washing the cells twice with phosphate-buffered saline and resuspending the cells to an OD600 of 0.4 in phosphate-buffered saline. OD measurements were calibrated against microscopic estimation of total length of filamentous cells (data not shown). Here, 200 μl of the resulting cell preparations (∼10 cm of cells) was added, in triplicate, to 5 g of site soil in a 38 ml serum bottle sealed with a rubber septum. Accompanying treatments consisted of (i) an uninoculated control (soil+200 μl phosphate-buffered saline) and (ii) a sterile control (soil+200 μl of the 10% Sabouraud dextrose-grown cell inoculum, sterilized by autoclaving). Next, 200 μl of filter-sterilized, aqueous 13C-phenol was added to each serum bottle resulting in a final soil concentration of 200 p.p.m. and an 8% soil moisture content. The dosed microcosms were stirred using a sterile spatula to ensure even distribution of added cells and labeled substrate. Bottles were then crimp sealed and incubated at 20 °C for 3 days. Headspace was analyzed by gas chromatography–mass spectrometry for 13CO2 production (Jeon et al., 2003; Padmanabhan et al., 2003; DeRito et al., 2005).
HPLC analysis of phenol
Phenol was analyzed by HPLC. Samples (1.0 ml) of culture medium were collected at various time points, immediately diluted with an equal volume of methanol, sealed and stored in screw-capped glass vials at 4 °C until analyzed. Samples were filtered through nylon acrodisc filters (0.2 μm; Acrodisc 25 mm syringe filter; Gelman, Ann Arbor, MI, USA). Phenol was separated using a Varian Microsorb-MV 100-5 C18 HPLC column (250 mm × 4.6 mm). A Waters model 590 HPLC pump was used to pump a mobile phase of methanol:40 mM acetic acid (60:40) at a flow rate of 1.0 ml min−1 (DeRito et al., 2005). Eluents were monitored by UV–VIS detection (ABI Analytical Absorbance Detector, Spectroflow 757) at a wavelength of 270 nm and quantified using external standard calibration curves.
Microscopy
A Nikon Eclipse E600 phase-contrast microscope equipped with a Hamamatsu color-chilled 3CCD camera (model C5810) was used to examine the isolates and to carry out direct cell counts. Images were collected using the software ATI Multimedia Center version 7.5 (ATI Technologies Inc., Sunnyvale, CA, USA). Aliquots of liquid culture (8 μl) were dispensed onto glass slides and Sabouraud dextrose agar slides (Murray et al., 1994). Slide cultures were incubated in the dark at room temperature for 24–48 h and then covered with glass cover slips prior to analysis.
Nucleotide sequence accession numbers
The nucleotide sequence data reported here have been submitted to GenBank under accession nos. EU675972–EU675994.
Results
The methodologies established in our previous study (DeRito et al., 2005) sought to identify soil microbial populations in the domain Bacteria involved in phenol degradation at an agricultural field site. These populations included (i) unenriched, primary degraders, (ii) enriched, primary degraders and (iii) trophically related organisms (carbon cross-feeders). In this study, DNA centrifugation gradients from our previous investigation were examined for fungal populations. PCR primers, originally designed for qualitative analysis of fungal communities by F-ARISA (fungal-automated rRNA intergenic spacer analysis) (Sequerra et al., 1997; Fisher and Triplett, 1999; Ranjard et al., 2001; Hansgate et al., 2005), but lacking the fluorescence label, were used to amplify sediment-derived 13C-DNA from five key soil treatments (Table 1; DeRito et al., 2005). Compared with treatments that received unlabeled phenol and only one dose of 13C-phenol, we obtained robust amplification of the targeted 18S–28S ITS gene region from diluted DNA extracted from the treatment which received multiple doses of 13C-phenol (Supplementary Figure 1, lanes 17–20). To avoid (minimize) the possibility of mistaking spurious PCR amplicons from SIP (that may result from background nucleic acid contamination in reagents) for 13C-labeled DNA, we cloned from dilutions from the 13C treatments that failed to yield amplicons in the corresponding 12C control treatment (DeRito et al., 2005). Additional steps for validating SIP results include isolation of microbial cultures, the genotype and phenotype of which match those found with SIP (see below).
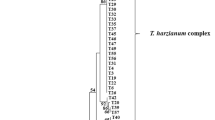
We prepared a clone library (a total of 80 white colonies) from the 13/13 field treatment-derived 18S–28S ITS amplicon. RFLP analysis of the resulting clone library revealed 15 patterns. Representatives of these were sequenced and compared with members of the NCBI database (http://www.ncbi.nlm.nih.gov/BLAST). The resulting dendrogram, which includes amplicon sizes for each clone, is shown in Figure 1. DNA sequences retrieved from the clone library were dominated by those identical to (100% sequence similarity) a member of the fungal species Trichosporon dulcitum from the NCBI database—41% of the clones analyzed shared identical RFLPs. In total, 3% of the clones fell within a closely related clade, T. multisporum (Figure 1). Other organisms represented in this dendrogram included members of the genera Cryptococcus, Rhodotorula, Nectria, Fusarium, Ascomycete and Acremonium. Because several prior studies that have found Trichosporon and other fungi to have phenol-degrading capacities (Neujahr and Varga, 1970; Rubin and Schmidt, 1985; Alexievaa et al., 2004; Margesin et al., 2005), we suspected that some, if not all, of the fungi identified in the 13/13 treatment may be primary degraders.

Phylogenetic analysis of cloned fungal genes (partial 18S rRNA, complete internal transcribed spacer region 1 (ITS1), 5.8S rRNA and ITS2, and partial 28S rRNA) from the sediment-derived 13C-DNA fraction of the 13/13 treatment. Clones were screened by restriction fragment length polymorphism (RFLP) and 21 were sequenced. Phylogenetic relationships were completed using the computational tools of DNASTAR Inc. and ClustalX version 1.83. Numbers at nodes are bootstrap values based on a neighbor-joining bootstrap analysis with 1000 replicates. Numbers in parentheses ( ) indicate the amplicon size for each clone in base pairs. Numbers in brackets [ ] indicate the percentage of clones analyzed with identical RFLPs (for example, 41% of the clones had identical RFLPs and are most closely identified with the species Trichosporon dulcitum. Rhizopus stolonifer is the outgroup.
To address this hypothesis, we attempted to grow and isolate phenol-degrading fungi from the site soil samples. After plating a dilution series (10−2–10−6) of a phenol-enrichment culture on minimal salts medium containing 250 p.p.m. phenol, 95 white filamentous colonies appeared on the 10−2 plate after 2 days. All colonies observed at each dilution were phenotypically indistinguishable. Next, 15 isolates were randomly selected and their ITS DNA was amplified by PCR and screened by RFLP. All 15 amplicons produced identical RFLP patterns, thus confirming population homogeneity (data not shown). Restreaking of two isolated colonies (on both yeast extract–peptone–galactose and Sabouraud dextrose agar plates) yielded several white/cream-colored, cone-shaped, wrinkled colonies. Microscopic observation of cultures grown on Sabouraud dextrose agar slides showed the presence of pseudohyphae and true hyphae (Figure 2)—characteristics of the genus Trichosporon (Kurtzman, 1988; Barnett et al., 2000). Moreover, the ITS sequence of our isolate was identical to that of a clone from our SIP-based library, confirming it to be a member of the species T. multisporum (Figure 1). One of these isolates (CD1) was examined for its ability to degrade phenol. HPLC analysis combined with spectrophotometric analysis showed that the isolate was able to metabolize and grow on 200 p.p.m. phenol within 24 h at 20 °C (Supplementary Figure 2).

Phase-contrast images of Trichosporon multisporum strain CD1. (a) Sabouraud dextrose agar slide culture and (b) Sabouraud dextrose broth culture. Note the presence of pseudohyphae and true hyphae (characteristics of the genus Trichosporon) in both (a, b).
We used a laboratory study to confirm that T. multisporum's ability to metabolize phenol could be expressed in the same soil used in the field SIP experiment (Figure 3). Phenol mineralization was dramatically accelerated in laboratory soil incubations in treatments inoculated with ∼10 cm of T. multisporum strain CD1 cells. In treatments receiving inocula, 30% of the added 13C label was recovered as 13CO2 within 24 h compared with the 2% recovered in uninoculated treatments (Figure 3). Subsequently, 11% of the added 13C label was recovered in the uninoculated control after 70 h. Thus, the ability of our isolate to grow on phenol is consistent with its role as being active in situ in phenol assimilation in soil.

Evolution of 13CO2 from [13C]phenol added to soil (final soil concentration of 200 p.p.m.) in laboratory incubations with and without Trichosporon multisporum inoculation. Treatments included addition of ∼2 cm cells g−1 T. multisporum strain CD cells grown on 0.4% glucose (Glu) or 10% Sabouraud dextrose (SD) and uninoculated and autoclaved controls. Net 13CO2 reflects total 13CO2 minus background 13CO2 (Padmanabhan et al., 2003). The percentages in parentheses indicate proportions of the total added [13C]carbon recovered as 13CO2. Data points are the averages of the results from three replicate treatments. Error bars indicate standard deviations.
Discussion
In this study, DNA-based SIP of the 18S–28S ITS rRNA gene region allowed us to identify a number of putative phenol-degrading fungi (basidiomycetes and ascomycetes) at our agricultural field site. We were able to discern from our electrophoresis gels, cloning, RFLP assays and sequencing procedures that the probed fungal community contained at least 12 different sized ITS1–5.8S–ITS2 amplicons, ranging from 516 to 629 bp (Figure 1). These lengths are within the range of those reported by Ranjard et al. (2001) for Basidiomycota and Ascomycota. The clone library produced from the PCR amplification of the ITS region contained members of the genera Trichosporon, Cryptococcus, Rhodotorula, Nectria, Fusarium, Ascomycete and Acremonium. Of these seven genera, isolated strains of Trichosporon, Cryptococcus, Rhodotorula and Fusarium have been shown to degrade phenol (Neujahr and Varga, 1970; Rubin and Schmidt, 1985; Margesin et al., 2003, 2005; Alexievaa et al., 2004; Atagana, 2004; Santos and Linardi, 2004; Bergauer et al., 2005; Krallish et al., 2006). Our attempt to isolate phenol-degrading fungi revealed only Trichosporon as a culturable, phenol-degrading yeast from our site. We speculate that the other sequences represented in our clone library were representative of either: (i) phenol degraders that failed to grow on our culture medium, (ii) secondary consumers in a phenol food web or (iii) spurious nucleic acid contaminants of the DNA-processing reagents. We took steps aimed at guarding against the latter possibility by cloning only from the pool of 13C-labeled DNA at a dilution that failed to yield a PCR amplicon in the corresponding 12C treatment (Padmanabhan et al., 2003; DeRito et al., 2005).
Similar to all procedures in environmental microbiology, SIP has strengths and weaknesses. The major strength is that SIP can find the ‘needle’ of rare, metabolically active populations in the inactive ‘haystack’ of a complex microbial community (Madsen, 2005). The major weakness of SIP derives from the various obstacles that must be overcome to accurately interpret resulting data. Discussed at length elsewhere (for example, Manefield et al., 2002a, 2002b; Lueders et al., 2004; Buckley et al., 2006; Madsen, 2006; Neufeld et al., 2007a, 2007b), these obstacles include the potential for both carbon cross-feeding (DeRito et al., 2005; Neufeld et al., 2007a) and erroneous, reagent-borne, false-positive sequences that may occur in clone libraries of the 13C-enriched nucleic acids. As discussed by Madsen (2006), routinely implemented SIP quality control procedures include short exposure to the labeled substrates (to minimize cross-feeding) and accounting for potential background contamination within the density gradients used to separate labeled from unlabeled nucleic acids. SIP procedures are successful mainly when the microbial populations that attack the labeled substrate also fully assimilate it. In many food chains, especially anaerobic ones (for example, Schink, 1997; Schmitz et al., 2006), the primary degraders of the added 13C-substrate may be so far removed from the final C-assimilation step that dilution of the labeled atoms may render isopycnic separation ineffective in identifying active populations (Kunapuli et al., 2007). Additional well-recognized sources of potentially misleading information include: PCR bias in preparing the clone libraries (for example, Suzuki and Giovannoni, 1996; von Wintzingerode et al., 1997) and small, incompletely sampled clone libraries that may not fully represent community members (for example, Hughes et al., 2001; Schloss et al., 2004; Tringe et al., 2005; Schloss and Handelsman, 2006). A robust strategy to assure the validity of SIP-derived sequence information is to succeed in isolating a microbial culture representative of the implicated sequence, and then to verify both that the cultured microorganism possesses the expected physiological capabilities and that it is active in its native habitat (Jeon et al., 2003; Madsen, 2005; Kasai et al., 2006; Pumphrey and Madsen, 2008). We pursued this strategy in the present study.
One of our isolates, T. multisporum strain CD1, was able to metabolize 200 p.p.m. within 24 h in broth culture. This finding is consistent with a previous study in which an isolated species of Trichosporon (T. cutaneum) showed visible growth on 500 p.p.m. phenol within 18 h (Neujahr and Varga, 1970). The ITS sequence of isolate CD1 was identical to that of an organism identified in our SIP experiment (clone F18; Figure 1). The resulting dendrogram revealed two distinct clades of Trichosporon. It may be noteworthy that isolate CD1 did not cluster with the dominant clade (41% of the library) of Trichosporon clones F2, F5 and F11. The ITS sequences of these clones were close (97.8%), but not identical to that of isolate CD1. Possible explanations for failing to cultivate representatives of the dominant clade include: (i) a relatively small number of isolates were sequenced (CD1 and CD2)—perhaps an isolate represented by clones F2, F5 and F11 was overlooked or (ii) organisms similar to clones F2, F5 and F11 were not able to flourish on the chosen agar medium.
To strengthen the case that T. multisporum strain CD1 was a key, primary phenol degrader in the field-based study, we implemented a laboratory-based study in which phenol mineralization was measured in site soil, both with and without added T. multisporum cells. We intentionally avoided physiologically biasing T. multisporum CD1 toward phenol metabolism by preparing the inocula from cultures grown on either 0.4% glucose or 10% Sabouraud dextrose. Results clearly showed that T. multisporum strain CD1 rapidly accelerated phenol mineralization in soil derived from its native habitat, thus strengthening our hypothesis that strain CD1's initial identification by SIP was the direct result of 13C assimilation from 13C-phenol. The data here provide new clues about Trichosporon's nutritional niche in nature and are consistent with prior reports (for example, Middelhoven et al., 2001) suggesting that Trichosporon actively participates in the mineralization of decaying plant material. In future studies, we plan to use T. multisporum in experiments aimed at exploring differential fitness traits between fungi and bacteria in the metabolism of carbon substrates in the soil habitat (de Boer et al., 2005).
Accession codes
References
Agency for Toxic Substances and Disease Registry (ATSDR) (2006). Toxicological Profiles: Phenol. US Agency for Toxic Substances and Disease Registry: Washington, DC.
Alexievaa Z, Gerginova M, Zlateva P, Paneva N . (2004). Comparison of growth kinetics and phenol metabolizing enzymes of Trichosporon cutaneum R57 and mutants with modified degradation abilities. Enzyme Microbiol Tech 34: 242–247.
Atagana HI . (2004). Biodegradation of phenol, o-cresol, m-cresol and p-cresol by indigenous soil fungi in soil contaminated with creosote. World J Microbiol Biotech 20: 851–858.
Barnett JA, Payne RW, Yarrow D . (2000). Yeasts: Characteristics and Identification, 3rd edn. Cambridge University Press: Cambridge, UK.
Bergauer P, Fonteyne PA, Nolard N, Schinner F, Margesin R . (2005). Biodegradation of phenol and phenol-related compounds by psychrophilic and cold-tolerant alpine yeasts. Chemosphere 59: 909–918.
Buchan A, Newell SY, Butler M, Biers EJ, Hollibaugh JT, Moran MA . (2003). Dynamics of bacterial and fungal communities on decaying salt marsh grass. Appl Environ Microbiol 69: 6676–6687.
Buckley DH, Huangyutitham V, Hsu SF, Nelson TA . (2006). Stable isotope probing achieved by disentangling the effects of genome G+C content and isotope enrichment on DNA density. Appl Environ Microbiol 73: 3189–3295.
Cullings KW, Vogler DR . (1998). A 5.8S nuclear ribosomal RNA gene sequence database: applications to ecology and evolution. Mol Ecol 7: 919–923.
Dagley S . (1967). The microbial metabolism of phenolics. In: McLaren AD, Peterson GH (eds). Soil Biochemistry, vol. 1. Marcel Dekker: New York. pp 287–317.
de Boer W, Folman LB, Summerbell RC, Boddy L . (2005). Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev 29: 795–811.
DeRito CM, Pumphrey GM, Madsen EL . (2005). Use of field-based stable isotope probing to identify adapted populations and track carbon flow through a phenol-degrading soil microbial community. Appl Environ Microbiol 71: 7858–7865.
Dumont MG, Murrell JC . (2005). Stable isotope probing—linking microbial diversity to function. Nat Rev Microbiol 3: 499–504.
Fisher MM, Triplett EW . (1999). Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl Environ Microbiol 65: 4630–4636.
Gaal A, Neujahr HY . (1979). Metabolism of phenol and resorcinol in Trichosporon cutaneum. J Bacteriol 137: 13–21.
Goerlitz DF, Troutman DE, Godsy EM, Franks BJ . (1985). Migration of wood-preserving chemical in contaminated groundwater in a sand aquifer at Pensacola, Florida. Environ Sci Technol 19: 955–961.
Hansgate AM, Schloss PD, Hay AG, Walker LP . (2005). Molecular characterization of fungal community dynamics in the initial stages of composting. FEMS Microbiol Ecol 51: 209–214.
Harris G, Ricketts RW . (1962). Metabolism of phenolic compounds by yeast. Nature 195: 473–474.
Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM . (2001). Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol 67: 4399–4406.
Jehmlich N, Schmidt F, von Bergen M, Richnow HH, Vogt C . (2008). Protein-based stable isotope probing (protein-SIP) reveals active species within anoxic mixed cultures. ISME J 2: 1122–1133.
Jenkins PG (ed). (1994). Environmental Health Criteria 161: Phenol. International Programme on Chemical Safety. World Health Organization: Geneva.
Jeon CO, Park W, Padmanabhan P, DeRito C, Snape JR, Madsen EL . (2003). Discovery of a bacterium, with distinctive dioxygenase, that is responsible for in situ biodegradation in contaminated sediment. Proc Natl Acad Sci USA 100: 13591–13596.
Jiang Y, Wen J, Lan L, Hu Z . (2007). Biodegradation of phenol and 4-chlorophenol by the yeast Candida tropicalis. Biodegradation 18: 719–729.
Jones MD, Singleton DR, Carstensen DP, Powell SN, Swanson JS, Pfaender FK et al. (2008). Effect of incubation conditions on the enrichment of pyrene-degrading bacteria identified by stable-isotope probing in aged, PAH-contaminated soil. Microbiol Ecol 56: 341–349.
Kasai Y, Takahata Y, Manefield M, Watanabe K . (2006). RNA-based stable isotope probing and isolation of anaerobic benzene-degrading bacteria from gasoline contaminated groundwater. Appl Environ Microbiol 72: 3586–3592.
Krallish I, Gonta S, Savenkova L, Bergauer P, Margesin R . (2006). Phenol degradation by immobilized cold-adapted yeast strains of Cryptococcus terreus and Rhodotorula creatinivora. Extremophiles 10: 441–449.
Kunapuli U, Lueders T, Meckenstock R . (2007). The use of stable isotope probing to identify key iron-reducing microorganisms involved in anaerobic benzene degradation. ISME J 1: 643–653.
Kurtzman CP . (1988). Identification and taxonomy. In: Kirsop BE, Kurtzman CP (eds). Yeasts: Living Resources for Biotechnology. Cambridge University Press: Cambridge, UK. pp 99–140.
Leigh MB, Pellizari VH, Uhlik O, Sutka R, Rodrigues J, Ostrom NE et al. (2007). Biphenyl-utilizing bacteria and their functional genes in a pine root zone contaminated with polychlorinated biphenyls (PCBs). ISME J 1: 134–148.
Lueders T, Wagner B, Claus P, Friedrich MW . (2004). Stable isotope probing of rRNA and DNA reveals a dynamic methylotroph community and trophic inter-actions with fungi and protozoa in oxic rice field soil. Environ Microbiol 6: 60–72.
Madsen EL . (2005). Identifying microorganisms responsible for ecologically significant biogeochemical processes. Nat Rev Microbiol 3: 439–446.
Madsen EL . (2006). The use of stable isotope probing techniques in bioreactor and field studies on bioremediation. Curr Opin Biotechnol 17: 92–97.
Manefield M, Whiteley AS, Griffiths RI, Bailey MJ . (2002a). RNA stable isotope probing, a novel means of linking microbial community function to phylogeny. Appl Environ Microbiol 68: 5367–5373.
Manefield M, Whiteley AS, Ostle N, Ineson P, Bailey MJ . (2002b). Technical considerations for RNA-based stable isotope probing: an approach to associating microbial diversity with microbial community function. Rapid Commun Mass Spectrom 16: 2179–2183.
Margesin R, Fonteyne PA, Redl B . (2005). Low-temperature biodegradation of high amounts of phenol by Rhodococcus spp. and basidiomycetous yeasts. Res Microbiol 156: 68–75.
Margesin R, Gander S, Zacke G, Gounot AM, Schinner F . (2003). Hydrocarbon degradation and enzyme activities of cold-adapted bacteria and yeasts. Extremophiles 7: 451–458.
Middelhoven WJ, Scorzetti G, Fell JW . (2001). Trichosporon porosum comb. nov., an anamorphic basidiomycetous yeast inhabiting soil, related to the loubiere/laibachii group of species that assimilate hemicelluloses and phenolic compounds. FEMS Yeast Res 1: 15–22.
Murray RGE, Doetch RN, Robinow CF . (1994). Determinative and cytological light microscopy. In: Gerhardt P, Murray RGE, Wood WA, Krieg NR (eds). Methods for General and Molecular Bacteriology. American Society for Microbiology: Washington, DC. pp 21–40.
Neufeld JD, Dumont MG, Vohra J, Murrell JC . (2007a). Methodological considerations for the use of stable isotope probing in microbial ecology. Microbiol Ecol 53: 435–442.
Neufeld JD, Vohra J, Dumont MG, Lueders T, Manefield M, Friedrich MW et al. (2007b). DNA stable-isotope probing. Nat Protoc 2: 860–866.
Neufeld JD, Wagner M, Murrell JC . (2007c). Who eats what, where and when? Isotope-labelling experiments are coming of age. ISME J 1: 103–110.
Neujahr HY, Varga JM . (1970). Degradation of phenols by intact cells and cell-free preparations of Trichosporon cutaneum. Eur J Biochem 13: 37–44.
Padmanabhan P, Padmanabhan S, DeRito C, Gray A, Gannon D, Snape JR et al. (2003). Respiration of 13C-labeled substrates added to soil in the field and subsequent 16S rRNA gene analysis of 13C-labeled soil DNA. Appl Environ Microbiol 69: 1614–1622.
Pumphrey GM, Madsen EL . (2008). Field-based stable isotope probing reveals the identities of benzoic acid-metabolizing microorganisms and their in situ growth in agricultural soil. Appl Environ Microbiol 74: 4111–4118.
Radajewski S, McDonald IR, Murrell JC . (2003). Stable-isotope probing of nucleic acids: a window to the function of uncultured microorganisms. Curr Opin Biotechnol 14: 296–302.
Ranjard L, Poly F, Lata JC, Mougel C, Thioulouse J, Nazaret S . (2001). Characterization of bacterial and fungal soil communities by automated ribosomal intergenic spacer analysis fingerprints: biological and methodological variability. Appl Environ Microbiol 67: 4479–4487.
Redecker D, Hijri M, Dulieu H, Sanders IR . (1999). Phylogenetic analysis of a dataset of fungal 5.8S rDNA sequences shows that highly divergent copies of internal transcribed spacers reported from Scutellospora castanea are of ascomycete origin. Fungal Genet Biol 28: 238–244.
Rubin HE, Schmidt S . (1985). Growth of phenol-mineralizing microorganisms in fresh water. Appl Environ Microbiol 49: 11–14.
Santos VL, Linardi VR . (2004). Biodegradation of phenol by a filamentous fungi isolated from industrial effluents—identification and degradation potential. Process Biochem 39: 1001–1006.
Schink B . (1997). Energetics of syntrophc cooperation in methanogenic degradation. Microbiol Molec Biol Rev 61: 262–280.
Schloss PD, Handelsman J . (2006). Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72: 6773–6779.
Schloss PD, Larget BR, Handelsman J . (2004). Integration of microbial ecology and statistics: a test to compare gene libraries. Appl Environ Microbiol 70: 5485–5492.
Schmitz RA, Daniel R, Deppenmeir U, Gottschalk G . (2006). The anaerobic way of life. In: Dworkin MW, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (eds). The Prokaryotes, vol. 2, 3rd edn. Springer Verlag: New York. pp 86–101.
Schwartz E . (2007). Characterization of growing microorganisms in soil by stable isotope probing with H218O. Appl Environ Microbiol 73: 2541–2546.
Scow HD, Goyer M, Payne E, Perwak J, Thomas R, Wood M . (1981). An Exposure and Risk Assessment for Phenol. Prepared for U.S. Environmental Protection Agency, Office of Water Regulations and Standard: Washington, DC, NTIS PB85-221695. pp 114–116.
Sequerra J, Marmeisse R, Valla G, Normand P, Capellano A, Moiroud A . (1997). Taxonomic position and intraspecific variability of the nodule forming Penicillium nodositatum inferred from RFLP analysis of the ribosomal intergenic spacer and random amplified polymorphic DNA. Mycol Res 101: 465–472.
Shoda M, Maruta K, Udaka S . (1980). Isolation and properties of phenol utilizing microorganisms with special reference to catechol 1,2-oxygenase. Agric Biol Chem 44: 1841–1846.
Singh H . (2006). Fungal metabolism of phenols, chlorophenols, and pentachlorophenol. In: Mycoremediation: Fungal Bioremediation. John Wiley & Sons Inc.: Hoboken, NJ. pp 215–282.
Stoilova I, Krastanov A, Stanchev V, Daniel D, Gerginova M, Alexievaa Z . (2006). Biodegradation of high amounts of phenol, catechol, 2,4-dichlorophenol and 2,6-dimethoxyphenol by Aspergillus awamori cells. Enzyme Microbiol Technol 39: 1036–1041.
Suzuki MT, Giovannoni S . (1996). Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62: 625–630.
Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW et al. (2005). Comparative metagenomics of microbial communities. Science 308: 554–557.
von Wintzingerode FV, Goebel JB, Stackebrandt E . (1997). Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21: 213–229.
Whiteley AS, Thomson B, Lueders T, Manefield M . (2007). RNA stable-isotope probing. Nat Protoc 2: 838–844.
Acknowledgements
This research was supported by the NIEHS Grant no. 1-R21-ES012834.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Rights and permissions
About this article
Cite this article
DeRito, C., Madsen, E. Stable isotope probing reveals Trichosporon yeast to be active in situ in soil phenol metabolism. ISME J 3, 477–485 (2009). https://doi.org/10.1038/ismej.2008.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2008.122
Keywords
This article is cited by
-
Identification of oleaginous yeasts that metabolize aromatic compounds
Journal of Industrial Microbiology and Biotechnology (2020)
-
Soil microbial communities in cucumber monoculture and rotation systems and their feedback effects on cucumber seedling growth
Plant and Soil (2017)
-
Performance and microbial community composition in a long-term sequential anaerobic-aerobic bioreactor operation treating coking wastewater
Applied Microbiology and Biotechnology (2016)
-
Investigation of the microbial metabolism of carbon dioxide and hydrogen in the kangaroo foregut by stable isotope probing
The ISME Journal (2014)
-
Importance of soil organic matter for the diversity of microorganisms involved in the degradation of organic pollutants
The ISME Journal (2014)
