
Abstract
Apart from water, tea is the world’s most widely consumed beverage. Tea is produced in more than 50 countries with an annual production of approximately 4.7 million tons. The market segment for specialty tea has been expanding rapidly owing to increased demand, resulting in higher revenues and profits for tea growers and the industry. Accurate varietal identification is critically important to ensure traceability and authentication of premium tea products, which in turn contribute to on-farm conservation of tea genetic diversity. Using a set of single nucleotide polymorphism (SNP) markers developed from the expressed sequence tag (EST) database of Camilla senensis, we genotyped deoxyribonucleic acid (DNA) samples extracted from a diverse group of tea varieties, including both fresh and processed commercial loose-leaf teas. The validation led to the designation of 60 SNPs that unambiguously identified all 40 tested tea varieties with high statistical rigor (p<0.0001). Varietal authenticity and genetic relationships among the analyzed cultivars were further characterized by ordination and Bayesian clustering analysis. These SNP markers, in combination with a high-throughput genotyping protocol, effectively established and verified specific DNA fingerprints for all tested tea varieties. This method provides a powerful tool for variety authentication and quality control for the tea industry. It is also highly useful for the management of tea genetic resources and breeding, where accurate and efficient genotype identification is essential.
Similar content being viewed by others


Introduction
Tea is the most widely consumed beverage in the world next to water.1 With an annual global production of 4.72 million tons (http://faostat.fao.org/), tea represents a $40 billion-a-year industry, with significant expected growth in non-Asian regions (http://www.worldteanews.com/, 2013). The tea plant, Camellia sinensis (L.) O. Kuntze, is a woody evergreen species in the family Theaceae, and in the subgenus section Thea.2,3 Its putative center of origin is in an area of South-East Asia that includes south and southwest China, Indo-China and northeastern India.2–5 Although tea is thought to have been domesticated in China, the exact region where tea came under cultivation is not clear and the ancestry of the cultigens has not been identified.3,6
Commercial tea products are classified into different categories based on processing techniques, i.e., manner of fermentation and oxidization. The common categories include green tea, black tea, oolong tea, white tea, yellow tea and dark tea. Within each category, a large number of varieties are used in tea production, often with greatly differing quality. It is estimated that in different regions of China, several thousand tea varieties are being cultivated. In addition, growing conditions, cultivation practices and harvesting time also significantly influence the quality and post-harvest attributes of tea.7
In spite of the significant effects of genotype on tea qualities, efficient methods for varietal authentication in the tea value chain have not yet been developed. Numerous instrumental methods to authenticate tea varieties have been investigated, among which near-infrared spectroscopy (1988) has been studied the most. This rapid and non-invasive method was employed by numerous investigators for authentication studies of tea.8–12 However, while near-infrared spectroscopy can effectively evaluate many quality attributes, accurate varietal identification remains an unsolved problem when large numbers of genotypes need to be examined. In addition to near-infrared spectroscopy, deoxyribonucleic acid (DNA)-based methods have been applied to identify plant species from a large array of commercial tea products.13 Microsatellite markers have been used in tea variety identification.14–22 In addition, sequence-tagged sites and cleaved amplified polymorphic sequences were applied to tea varietal identification.23,24 However, to date, the application of DNA fingerprinting has been used only for the differentiation of varieties, which precludes verification of large numbers of varieties through exact genotype matching. Moreover, resolving genotyping results from different labs, even with the use of microsatellite markers, has not been straightforward. It is difficult to standardize data generated from different genotyping platforms, and comparison of data is further complicated, because the same allele may be binned differently. Therefore, the use of simple sequence repeat (SSR)-based fingerprints for tea authentication can lead to false conclusions.
Recent progress in technology for plant genomics has led to the escalation in use of single nucleotide polymorphism (SNP) markers in DNA fingerprinting.25 The most abundant class of polymorphisms in plant genomes,26,27 SNPs have many advantages that are leading to their use as marker of choice. Unlike SSR markers, DNA separation by size is not required to analyze SNPs, and an assay array format or microchips can be used to accurately determine their identities. Because SNPs are biallelic and codominant markers, the error rate in allele calling is much lower than with SSRs and quick, low-cost, multiplex genotyping techniques can be employed. These advantages have resulted in SNPs increasingly becoming the markers of choice for accurate genotype identification and in crop improvement. Using a nanofluidic system to analyze SNP markers, Fang et al.28 generated SNP fingerprint patterns for small quantities of DNA extracted from the seed coat of single cacao beans. Based on the SNP profiles, an assumed adulterant variety was unambiguously distinguished from the authentic beans by multilocus matching.
Camellia sinensis has a genome size of 4.0 Gb.29 Full genomic sequences of Camellia sinensis have not been developed; however, a substantial amount of transcriptome data and various expressed sequence tags (ESTs) have been developed from different tissues, including young roots, flower buds, immature seeds and roots.19,20,30–32 The publicly accessible EST databases offer a low-cost source for an effective first step in SNP discovery. The objectives of the present study were to develop SNP markers through the data mining of EST databases of tea plants and assess their potential application for tea varietal identification. The SNP resources reported herein represent the first study of EST-derived SNP validation in tea and demonstrate the utility of EST databases as an alternative approach for de novo SNP identification in species whose genome sequences are not yet available. These SNP markers, as well as the genotyping method, would be particularly useful for varietal authentication, germplasm management and tea breeding programs.
Materials and methods
Mining of putative SNPs from tea EST database
EST sequences of Camellia sinensis were obtained from the EST database at the NCBI GenBank (http://www.ncbi.nlm.nih.gov/). The FASTA-formatted files of EST sequences were downloaded and merged into a single dataset for data mining. The mRNA and cDNA sequences in the dataset were removed using the program EST-trimmer (http://pgrc.ipk-gatersleben.de/misa/download/est_trimmer.pl). Redundant entries were examined and excluded using the CD-HIT program with a 95% sequence similarity threshold. Putative EST-SNPs were detected using the QualitySNP program.33,34 Only clusters that included at least six EST sequences, with a confidence score over 4, were accepted. In order to meet the requirements and constraints for primer design, all candidates for SNP markers with less than 60 nucleotides between two neighboring SNPs, and with flanking sequences less than 100 nucleotides long, were removed. A subset of the identified SNP sequences was then chosen for design and manufacture of primers to assay for SNPs in tea plant.
Validation of putative SNPs
To evaluate the putative SNP markers for suitability of varietal identification, we used a nanofluidic genotyping system and validated the SNPs for 40 diverse tea varieties (Table 1). Leaf samples of these varieties were obtained from the Tea Research Institute, Chinese Academy of Agricultural Sciences, Hangzhou, China. Actively growing shoot and leaf samples from all varieties were harvested and dried in silica gel (Table 1).
DNA was extracted from dried tea leaves with the DNeasy® Plant Mini kit (Qiagen Inc., Valencia, CA, USA), which is based on the use of silica as an affinity matrix. The dry leaf tissue was placed in a 2-mL microcentrifuge tube with one ¼-inch ceramic sphere and 0.15 g garnet matrix (Lysing Matrix A; MP Biomedicals, Solon, OH, USA). The leaf samples were disrupted by high-speed shaking in a TissueLyser II (Qiagen Inc.) at 30 Hz for 1 min. Lysis solution (DNeasy® kit buffer AP1 containing 25 mg mL−1 polyvinylpolypyrrolidone), along with ribonuclease I, was added to the powdered leaf samples and the mixture was incubated at 65 °C, as specified in the kit instructions. The remainder of the extraction method followed manufacturer’s suggestions. DNA was eluted from the silica column with two washes of 50 μL Buffer AE, which were pooled, resulting in 100 μL DNA solution. Using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA), DNA concentration was determined by absorbance at 260 nm. DNA purity was estimated by the 260∶280 ratio and the 260∶230 ratio.
Ninety-six putative SNP sequences were submitted to the Assay Design Group at Fluidigm Corporation (South San Francisco, CA, USA) for design and manufacture of primers for a SNPtypeTM genotyping panel. The assays were based on competitive allele-specific polymerase chain reaction (PCR) and enable bi-allelic scoring of SNPs at specific loci (KBioscience Ltd, Hoddesdon, UK).35 The Fluidigm SNPtypeTM Genotyping Reagent Kit was used according to the manufacturer’s instructions. Using these primers, the isolated DNAs were subjected to Specific Target Amplification in order to enrich the SNP sequences of interest.36 Genotyping was performed on a nanofluidic 96.96 Dynamic ArrayTM IFC (Integrated Fluidic Circuit; Fluidigm Corp.). This chip automatically assembles PCR reactions, enabling simultaneous testing of up to 96 samples with 96 SNP markers. The use of a 96.96 Dynamic Array IFC for SNP genotyping of human samples was described by Wang et al.36 End-point fluorescent images of the 96.96 IFC were acquired on an EP1TM imager (Fluidigm Corp.). The data was analyzed with Fluidigm Genotyping Analysis Software.37
Data analysis
Informativeness of SNP markers
Key descriptive statistics for measuring the informativeness of these 96 SNP markers were calculated, including minor allele frequency, observed heterozygosity, expected heterozygosity, Shannon’s information index and inbreeding coefficient.38 The program GenAlEx 6.5 was used for computation.39,40 For genotype identification, pairwise multilocus matching was applied among individual samples using the same program. DNA samples that were fully matched at the genotyped SNP loci were declared the same genotype (or clones).
To assess the differentiation power of the SNP markers, we calculated the probability of identity (PID).41 We computed the PID among siblings, which was defined as the probability that two sibling individuals drawn at random from a population have the same multilocus genotype.41
The overall PID among siblings is the upper limit of the possible ranges of PID in a population, thus providing the most conservative number of loci required to resolve all individuals, including relatives.41 This can be computed using the following equation:
where pi is the frequency of the ith allele.38
Analysis of genetic relationship among varieties
Distance-based multivariate analysis was used to assess the relationship among the individual varieties. Pairwise genetic distances as defined by Peakall et al.42 were computed using the DISTANCE procedure implemented in GenAlEx 6.5.39,40 The same program was then used to perform Principal Coordinates Analysis (PCoA), based on the pairwise distance matrix. Both distance and covariance were standardized.
A model-based clustering algorithm implemented in the STRUCTURE software program was applied to the SNP data.43 This algorithm attempted to identify genetically distinct subpopulations based on allele frequencies. The admixture model was applied and the number of clusters (K-value), indicating the number of subpopulations the program attempted to find, was set from 1 to 10. The analyses were carried out without assuming any prior information about the genetic group or geographic origin of the samples. Ten independent runs were assessed for each fixed number of clusters (K), each consisting of 1×106 iterations after a burn-in of 2×106 iterations. The ΔK value was used to detect the most probable number of clusters and the computation was performed using the online program STRUCTURE HARVESTER.44,45 Of the 10 independent runs, the one with the highest ln Pr (X|K) value (log probability or log likelihood) was chosen and represented as bar plots.
Results
SNP discovery
A total of 202278 ESTs and mRNA nucleotide sequences were downloaded from NCBI (26 August 2013), after adapter removal, trimming and quality control, 124 647 higher quality sequences were selected. CAP3 program was used to assembly sequences into 20 934 contigs and 40 810 singlets with an average size of 4.005 sequences per contig under default parameter, among which putative SNPs were detected in 789 contigs using the QualitySNP program.34 All of these selected clusters included a minimum of six EST sequences. In total, we obtained 1786 putative EST-SNPs, of which, 1193 were transition types, including 672 C/T and 521 A/G. There were 593 transversion types, including 162 A/T, 142 A/C, 162 T/G and 127 C/G. To select high-quality SNPs for validation, candidate SNP sites with at least 60 bp before and after the site were filtered. We calculated the number of all sequences in a cluster and the number containing the SNP type in this cluster. We then selected 96 SNPs for validation by genotyping a test panel of tea varieties, including both clonal and seed populations. The flanking sequences and SNPs are listed as Supplementary Table 1.
Frequency of SNP markers and descriptive statistics
Out of the 96 genotyped SNP markers, 75 were successfully genotyped (Figure 1), while genotyping failed for 21 SNPs (21.9%), likely due to the sequence complexity or the presence of polymorphisms within flanking sequences. Among the successfully genotyped SNPs, 15 were monomorphic across the 40 tea varieties (i.e., only one SNP variant was identified in all individuals). These monomorphic markers likely resulted from errors in EST sequencing, which then led to incorrect identification of SNP. It is also possible that some of these SNPs may correspond to rare alleles that were not present in the test panel of tea varieties.

Call map view from the dynamic array IFC, displaying SNP fingerprints of genotyped tea varieties, shows the computer generated image of the genotype calls for each of the individual reaction chambers. Each column (vertical direction) represents data from one assay that correlated to the SNP genotyping assay loaded from each assay inlet.
A total of 60 polymorphic SNPs were retained for further analysis. These 60 SNPs were reliably scored across the validation panel, and thus were considered true SNPs (Table 2). The minor allele frequency of these SNPs ranged from 0.03 to 0.485 with an average of 0.267. The mean observed heterozygosity was 0.324, ranging from 0.023 to 0.970, whereas the mean expected heterozygosity was 0.354 ranging from 0.045 to 0.579 (Table 2).
Variety authenticity
Repeated tests, using three independently extracted DNAs from the same tea variety, showed that the genotyping result was highly consistent. The seven repeatedly genotyped varieties (each with three replications) always generated the same SNP profiles. Individual genotype matching (pairwise comparisons) based on the 60 SNP markers showed that each of the 40 varieties has a unique SNP profile (Table 3). The probability that two tea varieties will have the same genotype at the 60 SNP loci is approximately 1 in 100 000 for the tested tea varieties, as computed by the mutlilocus matching procedure implemented in GenAlex 6.5.40
Genetic relationship among tea varieties
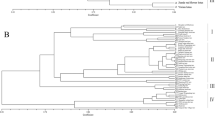
The genetic relationships among the 40 tea varieties are presented in the principal coordinates analysis plot (Figure 2). Each of the tested varieties was clearly separated from one another. Although the pattern of grouping was not apparent, it appeared that all the tested varieties could be grouped into three clusters. The first cluster was comprised mainly of the varieties from Southern China, including Guangxi and Guangdong. The second cluster included most of the varieties from Fujian Province, which is adjacent to Guangdong. The third cluster covered varieties from Zhejiang, Anhui and Jiangsu, including several of the well-known green tea varieties, such as ‘Longjing’ and ‘Bi Yun’.

PCoA plot of 40 tea varieties from Fujian, Guangdong, Guangxi, Anhui, Zhejiang and Jiangsu Provinces, China. The plane of the first three main PCO axes accounted for 43.4% of total variation. First axis=22.2% of total information, the second=11.6% and the third=9.6%. PCO, principle coordinate.
Population stratification of the 40 varieties, based on ΔK value computed by STRUCTURE HARVESTER, revealed two clusters as the most probable number of K44 (Figure 3) and the partition was largely compatible with the principle coordinate analysis (Figure 2). Almost all the varieties that fell into the cluster of Southern China (Guandong and Guangxi) in the PCoA were assigned to one Bayesian cluster. However, varieties which fell in the two clusters of Fujian and Eastern China in the PCoA were grouped in another single Bayesian cluster. Varieties ‘Shuigucha’ and ‘Yenhuayuanye’ were hybrids between the two clusters. This result appeared compatible with recent report based on SSR markers,46 where landraces from Southern China, and east coastal provinces (Zhejiang, Jiangsu and Anhui) were grouped in different clusters.

Inferred clusters in the Chinese tea varieties using STRUCTURE, where K is the potential number of genetic clusters that may exist in the overall sample of individuals. Each vertical line represents one individual multilocus genotype. Individuals with multiple colors have admixed genotypes from multiple clusters. Each color represents the most likely ancestry of the cluster from which the genotype or partial genotype was derived. Clusters of individuals are represented by colors.
Discussion
The market of specialty tea has been expanding rapidly at a global scale. So far, it is not possible to distinguish the genetic identity of a tea variety by other means, such as morphological and biochemical characteristics, especially for processed tea. Due to the insufficient throughput, accuracy and data standardization, the existing molecular marker-based technology, such as SSR marker fingerprinting, is of limited use. Tea leaf in commercial products usually contains a high level of polyphenolic and other PCR- inhibitory compounds and, in addition, there is residue from microorganisms that is a direct result of the fermentation and drying processes. Because of these problems, a robust analytical system is needed to genotype tea DNA.
In the present study, we demonstrated a DNA fingerprinting method that uses a small set of SNP markers to verify the genetic identity of a single bud or leaf. This method can handle a large amount of samples in a short period of time and the result is highly robust and repeatable. Our results showed that the nanofluidic array of SNP markers is particularly suitable for this purpose. The Specific Target Amplification protocol36 efficiently dealt with potential problems of the quality or quantity of DNA extracted from tea leaves. The Specific Target Amplification protocol, performed before genotyping, is a multiplex PCR reaction using primers for all loci of interest, but without targeting the specific alleles, thus proportionally increasing the copies of these loci. This procedure has been demonstrated to significantly improve call rates for SNPs in Chenopodium quinoa,47 Amaranthus,48 Rosoideae49 and cacao,28 and solved our problem of the low concentration of DNA obtained from processed commercial tea leaves. Results from the repeatedly genotyped leaves (three independent DNA extractions performed from the same tree) showed 100% concordance, suggesting that the nanofluidic system is a reliable platform for generating tea DNA fingerprints with high accuracy.
The effectiveness of individual identification via SNP fingerprints depends on the number of loci used for genotyping. An important statistical parameter for determining the number of loci required to identify all distinct individuals with a needed confidence level, is the PID. Multilocus PID values can be obtained by multiplying together single-locus PID values, assuming independence of loci. A stringent PID value is needed for domesticated crop species, because they often share similar ancestors. Therefore, PID calculated for sibs would provide a highly conservative boundary for domesticated crop species. The present result shows that using the 60 SNP loci, the chance of sampling identical genotypes from a random mating population would be 1 out 100 000. It thus predicts the high statistical power of using this set of SNPs for tea genotype verification.
Although the distance-based PCoA grouped the 40 varieties into three clusters, the Bayesian approach using STRUCTURE partitioned the tested varieties into two clusters (K=2). The varieties from Fujian were assigned together with those from eastern China in the Bayesian clustering analysis. If Fujian was included as part of eastern China, the result generated by PCoA would be fully compatible with the Bayesian clustering analysis. Among the varieties from Eastern China, ‘Zhenong 12’ showed a membership of Southern China. This is because ‘Zhenong 12’ is a hybrid variety of ‘Fuding Dabaicha’ and ‘Yunan Daye’, and none of these parents were traditional varieties from eastern China. The result is also compatible with the groupings reported in Yao et al.,46 which used microsatellite markers to assess genetic relationship among the Chinese tea varieties. In the report of Yao et al.,46 the assigned clusters of Chinese tea varieties largely followed the geographical distribution, indicating that there was substantial regional differentiation among the Chinese tea landraces. Even so, less than 50% of the total variation can be explained by the PCoA, indicating the variation between clusters (or between regions) is relatively small. This observation supported the conclusion of Yao et al.,46 which was based on SSR analysis, that most of the diversity was found within populations, whereas interpopulation variation accounted only for less than 5% of the total molecular variance. This result also supports previous reports that there might be a high level of inbreeding among the tested Chinese varieties, as indicated by a large inbreeding coefficient and reduced allelic diversity.46,50
There remain a large number of EST sequences available for tea, offering an attractive alternative resource for in silico SNP identification. More SNP markers will be needed in order to assess genetic relationship among different varieties. In addition, validation of tea EST-derived SNPs in a large number of tea germplasm groups will help the identification of fully informative SNPs, but also will highlight the proportion of informative SNPs shared across different germplasm groups. More stringent SNP quality control will be needed to minimize PCR artifacts. Moreover, multiple leaf samples should be included in genotyping to examine genotyping repeatability. Sample of internal control needs to be applied to detect possible inconsistency between different plates.
This information is crucial for selecting an optimum core set of SNP markers for tea varietal identification, as well as for assisting tea germplasm management and breeding.
In conclusion, we conducted a pilot study on varietal authentication for tea. We developed a set of SNP markers and used them for varietal genotyping using a nanofluidic array. This technology enabled us to generate high quality SNP profiles based on DNA extracted from both fresh and processed tea products, including white, green, oolong and black tea. Together with forensic statistical tools, these SNP-based DNA fingerprints allowed unambiguous identification of all tested varieties. To our knowledge, this is the first authentication study in commercial tea products using molecular makers. This approach is robust for authentication verification of specialty tea varieties and thus, has a significant potential for practical application.Footnote 1
Notes
Supplemental Information for this article can be found on the Horticulture Research website (http://www.nature.com/hortres).
References
Macfarlane A, Macfarlane I . The Empire of Tea. New York: The Overlook Press, 2004; 32.
Barua PK . Classification of the tea plant. Two Bud 1963; 10: 3–11.
Ming TL, Bartholomew B . Theaceae. In: Wu ZY, Raven PH, Hong DY, (eds.) Flora of China. Beijing/St Louis, MO: Science Press & Missouri Botanical Garden Press, 2007: 367.
Kingdon-Ward F . Does wild tea exist? Nature 1950; 4191: 297–299.
Chang HT, Bartholomew B . Camellias. Portland, OR: Timber Press, 1984.
Kulasegaram S . Progress in tea breeding. Trop Agric Res Ser 1978; 11: 151–160.
Heiss ML, Heiss RJ . The Story of Tea: A Cultural History and a Drinking Guide. Berkley, CA: Ten Speed Press, 2007: 417.
Luypaert J, Zhang MH, Massart DL . Feasibility study for the use of near infrared spectroscopy in the qualitative and quantitative analysis of green tea, Camellia sinensis (L.). Anal Chim Acta 2003; 478: 303–312.
Zhang MH, Luypaert J, Fernández Pierna JA et al. Determination of total antioxidant capacity in green tea by near-infrared spectroscopy and multivariate calibration. Talanta 2004; 62: 25–35.
Chen QS, Zhao JW, Liu MH et al. Determination of total polyphenols content in green tea using FT-NIR spectroscopy and different PLS algorithms. J Pharmaceut Biomed 2008; 46: 568–573.
He Y, Li XL, Deng XF . Discrimination of varieties of tea using near infrared spectroscopy by principal component analysis and BP model. J Food Eng 2007; 79: 1238–1242.
Tan SM, Luo RM, Zhou YP et al. Boosting partial least-squares discriminant analysis with application to near infrared spectroscopic tea variety discrimination. J Chemometr 2012; 26: 34–39.
Stoeckle MY, Gamble CC, Kirpekar R et al. Commercial teas highlight plant DNA barcode identification successes and obstacles. Sci Rep 2011; 1: 42.
Ujihara T, Ohta R, Hayashi N et al. Identification of Japanese and Chinese green tea cultivars by using simple sequence repeat markers to encourage proper labeling. Biosci Biotechnol Biochem 2009; 73: 15–20.
Ujihara T, Hayashi N, Tanaka J . Identification of material cultivar of green tea infusions by simple sequence repeat markers. Food Sci Technol Res 2012; 18: 209–217.
Kaundun SS, Matsumoto S . PCR-based amplicon length polymorphisms (ALPs) at microsatellite loci and indels from non-coding DNA regions of cloned genes as a means of authenticating commercial Japanese green teas. J Sci Food Agric 2004; 84: 895–902.
Chen L, Zhou ZX, Yang YJ . Genetic improvement and breeding of tea plant (Camellia sinensis) in China: from individual selection to hybridization and molecular breeding. Euphytica 2007; 154: 239–248.
Ma JQ, Zhou YH, Ma CL et al. Identification and characterization of 74 novel polymorphic EST-SSR markers in the tea plant, Camellia sinensis (Theaceae). Am J Bot 2010; 97: 153–156.
Sharma P, Kumar S . Differential display-mediated identification of three drought-responsive expressed sequence tags in tea [Camellia sinensis (L.) O. Kuntze]. J Bioscience 2005; 30: 231–235.
Taniguchi F, Fukuoka H, Tanaka J . Expressed sequence tags from organ-specific cDNA libraries of tea (Camellia sinensis) and polymorphisms and transferability of EST-SSRs across Camellia species. Breed Sci 2012; 62: 186–195.
Yang JB, Yang J, Li HT et al. Isolation and characterization of 15 microsatellite markers from wild tea plant (Camellia taliensis) using FIASCO method. Conserv Genet 2009; 10: 1621–1623.
Zhao LP, Liu Z, Chen L et al. Generation and characterization of 24 novel EST derived microsatellites from tea plant (Camellia sinensis) and cross-species amplification in its closely related species and varieties. Conserv Genet 2008; 9: 1327–1331.
Ujihara T, Taniguchi F, Tanaka J et al. Development of expressed sequence tag (EST)-based cleaved amplified polymorphic sequence (CAPS) markers of tea plant and their application to cultivar identification. J Agric Food Chem 2011; 59: 1557–1564.
Hu CY, Tsai YZ, Lin SF . Development of STS and CAPS markers for variety identification and genetic diversity analysis of tea germplasm in Taiwan. Bot Stud 2014; 55: 12.
Weising K, Nybom H, Pfenninger M et al. DNA Fingerprinting in Plants: Principles, Methods, and Applications. 2nd ed. Boca Raton, FL: CRC Press, 2005.
Buckler ES, Thornsberry J . Plant molecular diversity and applications to genomics. Curr Opin Plant Biol 2002; 5: 107–111.
Zhang DX, Hewitt GM . Nuclear DNA analyses in genetic studies of populations: practice, problems and prospects. Mol Ecol 2003; 12: 563–584.
Fang WP, Meinhardt LW, Mischke S et al. Accurate determination of genetic identity for a single cacao bean, using molecular markers with a nanofluidic system, ensures cocoa authentication. J Agric Food Chem 2014; 62: 481–487.
Tanaka J, Taniguchi F, Hirai N et al. Estimation of the genome size of tea (Camellia sinensis), camellia (C. japonica), and their interspecific hybrids by flow cytometry. Tea Res J 2006; 101: 1–7.
Chen J, Wang PS, Xia YM et al. Genetic diversity and differentiation of Camellia sinensis L. (cultivated tea) and its wild relatives in Yunnan province of China, revealed by morphology, biochemistry and allozyme studies. Genet Resources Crop Evol 2005; 52: 41–52.
Park JS, Kim JB, Hahn BS et al. EST analysis of genes involved in secondary metabolism inCamellia sinensis (tea), using suppression subtractive hybridization. Plant Sci 2004; 166: 953–961.
Shi CY, Yang H, Wei CL et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics 2011; 12: 131.
Li WZ, Godzik A . Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006; 22: 1658–1659.
Tang JF, Vosman B, Voorrips RE et al. QualitySNP: a pipeline for detecting single nucleotide polymorphisms and insertions/deletions in EST data from diploid and polyploid species. BMC Bioinformatics 2006; 7: 438.
McCouch SR, Zhao KY, Wright M et al. Development of genome-wide SNP assays for rice. Breeding Sci 2010; 60: 524–535.
Wang J, Lin M, Crenshaw A et al. High-throughput single nucleotide polymorphism genotyping using nanofluidic Dynamic Arrays. BMC Genomics 2009; 10: 561.
Fluidigm. Fluidigm SNP Genotyping User Guide Rev H1, PN 68000098. South San Francisco, CA: Fluidigm Corporation, 2011.
Evett IW, Weir BS . Interpreting DNA Evidence: Statistical Genetics for Forensic Scientists. Sunderland, MA: Sinauer, 1998.
Peakall R, Smouse PE . Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 2006; 6: 288–295.
Peakall R . Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 2012; 28: 2537–2539.
Waits LP, Luikart G, Taberlet P . Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 2001; 10: 249–256.
Peakall R, Smouse PE, Huff DR . Evolutionary implications of allozyme and RAPD variation in diploid populations of dioecious buffalograss Buchloe dactyloides. Mol Ecol 1995; 4: 135–147.
Pritchard JK, Stephens M, Donnelly P . Inference of population structure using multilocus genotype data. Genetics 2000; 155: 945–959.
Evanno G, Regnaut S, Goudet J et al. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 2005; 14: 2611–2620.
Earl DA, vonHoldt BM . STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resources 2012; 4: 359–361.
Yao MZ, Ma CL, Qiao TT et al. Diversity distribution and population structure of tea germplasms in China revealed by EST-SSR markers. Tree Genet Genomes 2012; 8: 205–220.
Maughan PJ, Smith SM, Rojas-Beltran JA et al. Single nucleotide polymorphism identification, characterization, and linkage mapping in quinoa. Plant Genome 2012; 5: 114–125.
Maughan PJ, Smith SM, Fairbanks DJ et al. Development, characterization, and linkage mapping of single nucleotide polymorphisms in the Grain Amaranths (Amaranthus sp.). Plant Genome 2011; 4: 92–101.
Longhi S, Giongo L, Buti M et al. Molecular genetics and genomics of the Rosoideae: state of the art and future perspectives. Hort Res 2014; 1: 1.
Zhao DW, Yang JB, Yang SX et al. Genetic diversity and domestication origin of tea plantCamellia taliensis (Theaceae) as revealed by microsatellite markers. BMC Plant Biol 2014; 14: 14.
Acknowledgements
We would like to give special thanks to Dr Liang Chen, The Tea Research Institute, Chinese Academy of Agricultural Sciences, for providing the tea germplasm samples and passport information and Stephen Pinney of USDA ARS for SNP genotyping of the tea samples. We would also like to thank Nico Tuason and the Assay design team for designing the SNP chip assay. This work was partially supported by the National Natural Science Foundation of China (30800884, 31370688). References to a company and/or product by the USDA are only for the purposes of information and do not imply approval or recommendation of the product to the exclusion of others that may also be suitable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Supplementary Table S1.
SNP markers and their flanking sequences for Camellia sinesis. (DOCX 25 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Fang, WP., Meinhardt, L., Tan, HW. et al. Varietal identification of tea (Camellia sinensis) using nanofluidic array of single nucleotide polymorphism (SNP) markers. Hortic Res 1, 14035 (2014). https://doi.org/10.1038/hortres.2014.35
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hortres.2014.35
This article is cited by
-
Genetic diversity and structure of Ugandan tea (Camellia sinensis (L.) O. Kuntze) germplasm and its implication in breeding
Genetic Resources and Crop Evolution (2024)
-
Quantitative detection of caffeine in beverages using flowing atmospheric-pressure afterglow (FAPA) ionization high-resolution mass spectrometry imaging and performance evaluation of different thin-layer chromatography plates as sample substrates
Analytical and Bioanalytical Chemistry (2022)
-
Genetic diversity of oolong tea (Camellia sinensis) germplasms based on the nanofluidic array of single-nucleotide polymorphism (SNP) markers
Tree Genetics & Genomes (2020)
-
Molecular Characterization of a Cacao Germplasm Collection Maintained in Yunnan, China Using Single Nucleotide Polymorphism (SNP) Markers
Tropical Plant Biology (2020)
-
Genetic diversity, linkage disequilibrium, and population structure analysis of the tea plant (Camellia sinensis) from an origin center, Guizhou plateau, using genome-wide SNPs developed by genotyping-by-sequencing
BMC Plant Biology (2019)
