
Abstract
Ichthyosis prematurity syndrome (IPS) is a rare autosomal recessive disorder characterized by prematurity, a thick caseous scale at birth and lifelong atopic diathesis. Here, we describe the first Japanese case of IPS and report novel compound heterozygous mutations (p.C403Y and p.R510H) in fatty acid transport protein 4 (FATP4). She is the first reported patient of Asian origin, entirely distinct from the Scandinavian population, in whom the heterozygote carrier frequency is very high.
Similar content being viewed by others
Ichthyosis prematurity syndrome (IPS) is a rare autosomal recessive disorder characterized by prematurity, respiratory distress and a thick caseous scale at birth.1 Life-threatening complications cease after the perinatal period. Skin findings change to a mild form of generalized ichthyosis, often associated with severe pruritus. Many patients also develop food/respiratory allergies with elevated IgE levels and eosinophilia. Defects in skin barrier function are thought to promote transcutaneous sensitization to both food and environmental allergens. These defects may also result in systemic responses such as increased total IgE and eosinophilia. These features are typically indicative of atopic diathesis.
Here, we present the case of a 14-year-old female with atopic dermatitis, increased total IgE and eosinophilia. This patient represents the first Japanese case of genetically diagnosed IPS. Using a next-generation sequencing strategy, we identified compound heterozygosity of novel missense mutations in the fatty acid transporter member 4 (FATP4) gene (p.C403Y and p.R510H).
The proposita was born at the Tosei General Hospital of nonconsanguineous parents. The birth occurred at 32 weeks, 2 days of gestation via spontaneous vaginal delivery. The birth weight was 2,186 g (>90 percentile). The patient had no family history of ichthyosis. Apgar scores were 2 and 4 at 1 and 5 min, respectively. Tracheal intubation was performed immediately. The patient was placed on a respirator with high inspiratory pressure and she received surfactant replacement therapy. Respiratory assistance was discontinued on day 3 with no further need of oxygen or positive-pressure breathing.
At birth, the patient was coated in a thick layer of white, vernix, caseosa-like material (Figure 1). The entire body, including the face, showed thickening and scaling of the skin, which became bright red during crying. Histological analysis of skin biopsy samples showed a diffuse hyperkeratosis with intracorneal pustule and psoriasiform hyperpalsia foci. No evidence of epidermolytic hyperkeratosis was recognized, resulting in a tentative diagnosis of nonbullous congenital ichthyosiform erythroderma. Transient eosinophilia up to 8,400/μl was noted during hospitalization, but this level gradually normalized. After discharge, the eruption improved into a mild flexural dermatitis with generalized ichthyosis under moisturizing-agent treatment.

Clinical skin features seen at birth.
When the patient was 14 years old, she visited a nearby clinic because of abdominal pain. Blood examination revealed eosinophilia and elevated serum IgE levels. She was referred to the Tosei General Hospital for further examination.
Thickening of the skin of the patient’s forehead, trunk and limbs was observed at the first visit. This thickening was accompanied by fine desquamation and mild pigmentation. The patient’s limbs were also slightly reddish. Blood examination revealed eosinophilia (2,068/μl), elevated serum IgE (18,000 IU/ml) and multiple sensitizations to respiratory and food allergens (specific IgE to house dust, 84.9 UA/ml; Dermatophagoides pteronyssinus, 93.5 UA/ml; cat dander 2.51 UA/ml; Japanese cedar, 2.21 UA/ml; egg white, 1.06 UA/ml; and ovomucoid, 0.91 UA/ml). As she had no past history of recurrent infections suggesting hyper IgE syndrome, atopic diathesis due to skin barrier defects was speculated.
As the list of differential diagnosis included a variety of nonbullous congenital ichthyosiform erythrodermas,2 we resorted to next-generation sequencing analysis. Targeted exome sequencing was performed using a Trusight Exome sequencing panel on a MiSeq platform (Illumina, San Diego, CA, USA). This kit enables enrichment and final analysis of a panel of ~2,700 genes. The patient and her parents gave written informed consent for genome analysis.
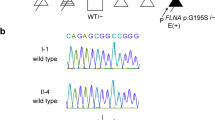
Of the nonsynonymous single-nucleotide variants (SNVs) detected in FATP4, two (c.G1208A and c.G1529A) were not found in the 1000 Genomes database (1000g2012feb), the Exome Variant Server database from 13,006 chromosomes (NHLBI GO Exome Sequencing Project)3 or the Japanese genetic variation database (Human Genetic Variation Database).4 These variations were strictly conserved among multiple species (Figure 2a) and were predicted to damage protein function by PolyPhen2 (scores 0.996 and 1, respectively) and AVSIFT (score 0 for both SNVs). Mutation validation was performed using PCR and Sanger sequencing of the candidate gene region. This analysis revealed that c.G1208A and c.G1529A were of paternal and maternal origins, respectively (Figure 2b).

(a) Conservation of the FATP4 protein near two SNVs. (b) Chromatogram derived from targeted capillary sequencing of the patient and her parents for mutations in FATP4, c.1208G>A and c.1529G>A. (c) Schematic drawing of the FATP4 protein and a summary of the mutations reported previously (black arrow) and in this report (red arrow). Functional domains include the transmembrane region (TM), an ER localization signal (ERx) and ATP/AMP (ATP/AMP) and very long-chain acyl-CoA synthetases/fatty acid transport proteins (VLACS/FATP) (FATP) motifs.
The FATP4 gene, consisting of 13 exons, produces a transmembrane protein that transports exogenous fatty acids into cells. The gene also functions as an acyl-CoA synthetase. FATP4 contains an N-terminal transmembrane region, an ER localization signal an AMP-binding domain involved in ATP binding and adenylate formation (ATP/AMP) and a conserved VLACS/FATP motif, which is important for fatty acid binding (VLACS; Figure 2c). IPS is very rare, except in Scandinavia with an estimated heterozygote carrier frequency of 1 in 50.1 Although 13 FATP4 mutations (2 nonsense, 9 missense and 2 splice site) have been reported to date (Figure 2c),5–10 all except two (one from the Middle East and the other from North Africa) are of European origin. Thus, p.C403Y and p.R510H are the first mutations reported in individuals of Asian origin. Clinical features in these individuals were not found to be substantially different from those of Scandinavian origin. Although IPS has not been reported in Japan, a lack of clinician experience may lead to potential cases going undiagnosed. Genetic testing can help to provide a correct diagnosis. Parents can then be informed of the prognostically favorable nature of IPS and its mode of inheritance. This information is necessary for genetic counseling.
Common loss-of-function mutations of the epidermal barrier protein filaggrin, the causative gene of ichthyosis vulgaris, are the major predisposing factors for atopic diathesis.11 Further study of IPS will contributes to our understanding of the relationship between skin barrier defects and atopic diathesis.
References
References
Klar J, Gedde-Dahl T Jr., Larsson M, Pigg M, Carlsson B, Tentler Ds et al. Assignment of the locus for ichthyosis prematurity syndrome to chromosome 9q33.3-34.13. J Med Genet 2004; 41: 208–212.
Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010; 63: 607–641.
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. URL: http://evs.gs.washington.edu/EVS/. Accessed December 2014.
Human Genetic Variation Browser. http://www.genome.med.kyoto-u.ac.jp/SnpDB.
Klar J, Schweiger M, Zimmerman R, Zechner R, Li H, Törmä H et al. Mutations in the fatty acid transport protein 4 gene cause the ichthyosis prematurity syndrome. Am J Hum Genet 2009; 85: 248–253.
Morice-Picard F, Léauté-Labrèze C, Décor A, Boralevi F, Lacombe D, Taieb A et al. A novel mutation in the fatty acid transport protein 4 gene in a patient initially described as affected by self-healing congenital verruciform hyperkeratosis. Am J Med Genet A 2010; 152A: 2664–2665.
Sobol M, Dahl N, Klar J . FATP4 missense and nonsense mutations cause similar features in ichthyosis prematurity syndrome. BMC Res Notes 2011; 4: 90.
Inhoff O, Hausser I, Schneider SW, Khnykin D, Jahnsen FL, Sartoris J et al. Ichthyosis prematurity syndrome caused by a novel fatty acid transport protein 4 gene mutation in a German infant. Arch Dermatol 2011; 147: 750–752.
Khnykin D, Rønnevig J, Johnsson M, Sitek JC, Blaas HG, Hausser I et al. Ichthyosis prematurity syndrome: clinical evaluation of 17 families with a rare disorder of lipid metabolism. J Am Acad Dermatol 2012; 66: 606–616.
Kiely C, Devaney D, Fischer J, Lenane P, Irvine AD . Ichthyosis prematurity syndrome: a case report and review of known mutations. Pediatr Dermatol 2014; 31: 517–518.
Irvine AD, McLean WH, Leung DY . Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011; 365: 1315–1327.
Data Citations
Tsuge, Ikuya HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.568 (2015)
Tsuge, Ikuya HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.570 (2015)
Acknowledgements
We thank Ms. Miyuki Teshigawara for her technical assistance. This work was supported by the MEXT-Supported Program for the Strategic Research Foundation at Private Universities from the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Tsuge, I., Morishita, M., Kato, T. et al. Identification of novel FATP4 mutations in a Japanese patient with ichthyosis prematurity syndrome. Hum Genome Var 2, 15003 (2015). https://doi.org/10.1038/hgv.2015.3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.3
