
Abstract
Population genetics theory predicts loss in genetic variability because of drift and inbreeding in isolated plant populations; however, it has been argued that long-distance pollination and seed dispersal may be able to maintain gene flow, even in highly fragmented landscapes. We tested how historical effective population size, historical migration and contemporary landscape structure, such as forest cover, patch isolation and matrix resistance, affect genetic variability and differentiation of seedlings in a tropical palm (Euterpe edulis) in a human-modified rainforest. We sampled 16 sites within five landscapes in the Brazilian Atlantic forest and assessed genetic variability and differentiation using eight microsatellite loci. Using a model selection approach, none of the covariates explained the variation observed in inbreeding coefficients among populations. The variation in genetic diversity among sites was best explained by historical effective population size. Allelic richness was best explained by historical effective population size and matrix resistance, whereas genetic differentiation was explained by matrix resistance. Coalescence analysis revealed high historical migration between sites within landscapes and constant historical population sizes, showing that the genetic differentiation is most likely due to recent changes caused by habitat loss and fragmentation. Overall, recent landscape changes have a greater influence on among-population genetic variation than historical gene flow process. As immediate restoration actions in landscapes with low forest amount, the development of more permeable matrices to allow the movement of pollinators and seed dispersers may be an effective strategy to maintain microevolutionary processes.
Similar content being viewed by others
Introduction
Population genetics theory predicts that detrimental effects of habitat loss and fragmentation will increase population genetic structure (Young et al., 1996), although many empirical studies in plants have not observed significant changes after habitat loss (e.g., Collevatti et al., 2001; Côrtes et al., 2013). Habitat loss and fragmentation may lead to marked reductions in population size and increase the spatial isolation of populations (Fahrig, 2003). The isolation of populations may raise inbreeding levels by increasing the probability of mating between closely related individuals and self-pollination because of, for example, changes in the composition and behavior of pollinators. Isolation may also limit the dispersal among populations, reducing gene flow and population connectivity (Young et al., 1996). This may cause disruption of the migration–drift equilibrium, because alleles lost by random genetic drift may not be rescued by gene flow (Young et al., 1996). Ultimately, the loss of genetic variability may reduce individual fitness and the ability to cope with environmental changes (Bijlsma and Loeschcke, 2012).
However, different factors can influence the detection of fragmentation effects on plant genetic structure, such as life history traits (e.g., mating system and pollination and seed dispersal modes) and time-lag effects (Collevatti et al., 2001; Kramer et al., 2008). For example, insect-pollinated plants are more likely to lose genetic variability because of fragmentation than bird-pollinated species because birds are more likely to move (and disperse pollen) over longer distances than insects (e.g., Kramer et al., 2011). Pollinators can also be adaptable and resilient to habitat loss and fragmentation (e.g., White et al., 2002), resulting in high gene flow and low genetic differentiation among plant populations as a consequence of high rates of pollen flow. Likewise, self-compatible plants may be less affected by habitat loss and fragmentation than obligate outcrossing plants (Aguilar et al., 2008).
Adults and seedlings can also show distinct responses to habitat loss and fragmentation (Van Geert et al., 2008). Genetic variability and differentiation of adults of perennial plants often show responses to past landscape conditions but not to recent habitat changes (Collevatti et al., 2001; Kramer et al., 2008). Nevertheless, several studies found decreasing genetic variability and increasing inbreeding coefficients in the progeny (seedlings), suggesting that there may be a time lag before ongoing habitat fragmentation is observed in the genetic structure of adults (Kettle et al., 2007; Aguilar et al., 2008; Van Geert et al., 2008). Therefore, assessing genetic variability in seedlings instead of adults in fragmented landscapes is useful when evaluating the ongoing effects of habitat loss and fragmentation on genetic variability and differentiation.
Although studies emphasize the effects of habitat loss and fragmentation on genetic variability, most of them compare patterns across sites or only focus on a given landscape (Keyghobadi et al., 2005a; Balkenhol et al., 2013). This approach, however, does not provide direct insights into the factors affecting genetic variability (Storfer et al., 2010). One effective way to address the impact of habitat loss and fragmentation on genetic variability and differentiation at the landscape scale is comparing multiple landscapes while taking into account the influence of landscape structure (Keyghobadi et al., 2005a; Storfer et al., 2010). This approach is particularly important in human-modified landscapes with high levels of biodiversity, such as the Atlantic forest in Brazil (Ribeiro et al., 2009).
The Atlantic forest has been reduced to <12% of its original 150 million ha (Ribeiro et al., 2009), resulting in islands of wild habitat surrounded by crops, pastures and urban matrix. Because of marked habitat loss, 84% of remnants are small (<50 ha), highly isolated (average distance between patches is 1440 m) and negatively influenced by edge effects (half of remaining forest is <100 m from the edge; Ribeiro et al., 2009). One of the dominant plant species of this biome is the keystone palm Euterpe edulis. Although once abundant, this palm species is currently endangered and locally extinct in many areas owing to illegal harvesting of the edible meristem (heart of palm; Galetti and Fernandez, 1998). E. edulis is a self-compatible monoecious species, but with predominant outcrossed reproduction (Gaiotto et al., 2003) and pollination performed mainly by small-sized bees (e.g., Trigona spinipes; Reis et al., 2000). Their fruits are eaten by more than 58 birds and 20 mammalian species but are dispersed mostly by large frugivorous birds (e.g., bellbirds Procnias nudicollis, toucans Ramphastos spp.) and thrushes (Turdus spp.; Galetti et al., 2013).
We analyzed the effect of historical effective population size and different landscape structure metrics on population genetic variability, inbreeding and genetic differentiation of E. edulis populations. Concomitantly, we also assessed the relative contribution of geographic distance to the variability of genetic differentiation between patches. Because plants and their mutualists (pollinators and seed dispersers) are likely to decline after loss of natural habitat and animals are less likely to cross unsuitable matrix habitats, our hypotheses are that inbreeding and genetic differentiation will be higher leading to reduced genetic variability in landscapes with lower forest amount, higher patch isolation and higher matrix resistance.
Materials and methods
Landscapes and sites selection
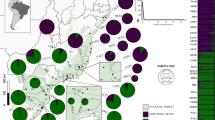
The study was carried out in five landscapes of the Atlantic forest in São Paulo state, Southeast Brazil (Figure 1a). The fragmentation of Atlantic forest in São Paulo dated from the nineteenth century, with the establishment of coffee plantations. Currently, the forest remnants are surrounded by different matrix types such as sugar cane, cattle pasture, forest plantations (Eucalyptus spp. and Pinus spp.) and coffee. The heterogeneous matrix is made of patches that can be more or less permeable to propagule movement, leading to high variation in the influence of matrix on the genetic variability and genetic differentiation among populations of the focal species.

(a) Atlantic forest remnants (gray) and the location of five sampled landscapes where E. edulis is present within São Paulo state, Southeast Brazil. (b) Representation of four of our sampled landscapes (2 km radius) illustrating the gradient of Atlantic forest cover (5, 20, 45 and 75%).
We sampled a total of 16 sites within the five landscapes distributed in a gradient of forest cover from 5 to 75% (Figure 1). Each landscape has two to four sites with populations of E. edulis (see Supplementary Information and Supplementary Table S1 for details). The sites can be distinct forest fragments in the case of very fragmented landscapes or more than one sampling position within the same fragment for landscapes containing large forest remnants (i.e. large forest reserves). Each landscape was defined by a circle of 2 km radius around a midpoint among the sites where E. edulis were sampled (Figure 1b). We chose this radius because foraging distance of potential pollinators, such as Plebleia droryana and Trigona spinipis, can reach 540 and 840 m, respectively (Zurbuchen et al., 2010) and seed dispersal distances by large bird species taxonomically related with the dispersers of E. edulis are likely to be shorter than 600 m (Holbrook, 2011). Using this experimental design, we were able to assess the effects of landscape structure and geographic distance between sites on genetic variability and genetic differentiation among sites within landscapes.
Seedling sampling
We sampled ~30 seedlings in each of the 16 sites (total of 463 individuals, see Supplementary Information and Supplementary Table S1). We used seedlings instead of adults to minimize the effect of time lag on genetic responses, because adult individuals may respond to past landscape structure (Kramer et al., 2008). To minimize the effects of spatial genetic autocorrelation, seedlings were sampled at 10 m of distance owing to significant spatial autocorrelation in relatedness below that distance (Carvalho, 2010). Leaf tissues were dried in silica gel and stored at −80 °C.
Genetic analysis
For genetic data analysis, genomic DNA extraction followed the CTAB (cetyltrimethyl ammonium bromide) procedure and all individuals were genotyped using eight highly polymorphic microsatellite loci (EE5, EE8, EE25, EE43, EE45, EE47, EE52 and EE63), following PCR protocol described by Gaiotto et al. (2001). DNA fragments were sized in ABI Prism 3100 automated DNA sequencer (Applied Biosystems, Foster City, CA, USA) using GeneScan ROX 500 size standard (Applied Biosystems), and were scored using GeneMaper v.4.1 software (Applied Biosystems). Ten percent of all individuals were genotyped two times in independent PCR amplifications to calculate genotyping error (alleles dropout and null alleles).
Genetic variability and genetic differentiation
The analyses of landscape structure effect on genetic parameters were performed at the node and link levels (see Wagner and Fortin, 2013). At the node level (genetic variability within each site), we estimated genetic diversity (He—expected heterozygosity under Hardy–Weinberg equilibrium, following Nei, 1978), AR based on rarefaction analysis (AR; Mousadik and Petit, 1996) and inbreeding coefficient (FIS, obtained from the analysis of variance of allelic frequency; Weir and Cockerham, 1984). Analyses were carried out using the software FSTAT 2.9.3.2 (Goudet, 2002). We also used the software INEST (Chybicki and Burczyk, 2009) to estimate a corrected FIS accounting for the potential impacts of null alleles. To conduct this analysis, we used the model based on a Bayesian approach (IIM), with 5 00 000 Markov chain Monte Carlo interactions and burn-in of 50 000.
At the link level, we estimated genetic differentiation among all pairs of sites nested within landscapes using three distinct measures: Wright’s FST, obtained from an analysis of variance of allele frequencies (Weir and Cockerham, 1984); GST' (Hedrick, 2005), based on FST, but taking into account observed diversity within population and number of sub-populations; and Jost's D (Jost, 2008), based on effective number of alleles instead of expected heterozygosity.
All measures have their advantages and drawbacks, for example, FST is influenced by heterozygozity; however, it is a widely used parameter and, as recommended by Meirmans and Hedrick (2011), should appear in all studies to allow comparisons with other studies. Jost's D is less suited for inferences about demography because it is insensitive to population size. One advantage of GST' is that it can be applied to every FST analog for which the maximum value can be obtained. Measures of genetic differentiation were calculated using the package ‘mmod’ (Winter et al., 2014) implemented in R software (R Development Core Team, 2013).
Because microsatellite markers may follow the stepwise mutation model, we estimated Slatkin's RST and tested the hypothesis that FST=RST using permutation test implemented in Spagedi (Hardy and Vekemans, 2002). A significant difference between RST and FST implies that stepwise-like mutations contributed to population differentiation and that, in that data set, RST performs better than FST to estimate population differentiation (Hardy et al., 2003).
Landscape mapping and explanatory covariates
We mapped the five landscapes using visual digitalization and manual classification at the scale of 1:5000 using high-resolution 1x1 m2 images available at Google Earth (http://earth.google.com). QGIS (http://www.qgis.org) software was used to access the images with the OpenLayer plugin (http://www.openlayers.org). Land use in each landscape was originally classified in 13 different classes: (i) advanced forest; (ii) intermediate forest succession; (iii) pioneer forest; (iv) initial forest regeneration; (v) Eucalyptus spp. plantation; (vi) sugar cane; (vii) coffee plantation; (viii) swamp; (ix) swamp with isolated trees; (x) pasture, (xi) pasture with isolated trees; (xii) mining areas and (xiii) rural household (see Supplementary Information and Supplementary Figure S1 for one example of land use classification and mapping).
For each analysis level (node and link), we calculated several landscape metrics related to forest amount, matrix resistance, site isolation and geographic distance. We performed Pearson’s correlation analysis to choose only biologically meaningful landscape metrics, excluding those with correlation coefficients <0.5.
At the node level, we calculated the percentage of forest cover in the vicinities of the focal site (search radius of 500 m) and patch size to represent forest amount. To calculate matrix resistance (the resistance that a landscape offers to pollinators and seed dispersers to disperse from one site to another), we used the software LORACS (Pinto et al., 2012). This software estimates the cost (matrix resistance) using multipatch (i.e. multiple corridors) biological flow based on graph theory. LORACS simulates multiple routes between two points (sampling sites) using a resistance surface, which was built by assigning resistance values to each land use type. These values represent the degree to which the land cover type facilitates or inhibits the movement and dispersal of individuals. We generated the resistance surface file for pollinators and seed dispersers of E. edulis. This was carried out using the approach proposed by Mühlner et al. (2010), based on expert opinion. Two ornithologists and one entomologist were interviewed about the potential resistance of different land uses to the movement of pollinators and seed dispersers, respectively (the list of pollinators and seed dispersers can be found in the Supplementary Information). Each expert independently attributed resistance values (from 0 to 5) to each type of land use, considering each pollinator and seed disperser. As we had the opinion of two ornithologists, we calculated Pearson’s correlation between the weights given by these experts to control the consistency in resistance classification. Correlations between the resistances of each land use type were high between the ornithologists (>0.6, see Supplementary Information and Supplementary Table S2), showing consistency between expert opinions. Thus, for the following analyses, we used the mean resistance value of each land use given by the ornithologists for each seed disperser. For the resistance value given by the entomologist, we used the raw data. We performed a principal component analysis with the data set of resistance value of each land use for each pollinator and seed disperser and ranked the value of the first principal component from 1 to 100 to obtain the final resistance weight of each land use type (Supplementary Information and Supplementary Table S3). We used the first principal component, which corresponds to 71% of the cumulative variance (see Supplementary Information and Supplementary Figure S16 for principal component analysis plot). From the analysis performed using the software LORACS, we obtained the cost of movement between pairs of sites within landscapes (multiple shortest path), which corresponds to the matrix resistance between pairs of sites. Because the source and target sites must be indicated for resistance analysis using the software LORACS, we calculated the mean matrix resistance of a selected site (source) in relation to all sites (target) to estimate the matrix resistance of one site.
To calculate site isolation, we used two landscape metrics: site isolation and proximity index. The site isolation was defined as the mean geographic distance, from border to border, between all paired sites within each landscape. The proximity index considers the size (m2) and proximity (m) of all sites in which a border exists within the search radius of focal site. This metric is calculated by summing the size of all sites whose borders are within the search radius of the focal site and dividing by the square of the distance from the focal site. It was calculated using the V-late extension within ArcGis 9.3, with a searching radius of 2000 m.
At the link level, we calculated the forest amount drawing a buffer of 500 m around a straight line between paired sites and calculating the percentage of forest within the buffer. To estimate the matrix resistance between sites, we followed a similar protocol used for node level but estimating the matrix resistance between site A (source) and site B (target) and between site B (source) and A (target), and calculated the mean matrix resistance between sites. Geographic distances between all site pairs within each landscape were calculated using Euclidean distance (in m), from border to border.
Many landscape metrics presented high correlation with each other (see Supplementary Information, Supplementary Tables S4 and S5). For example, patch size metrics were not used as a landscape feature in our model because of high correlation with other landscape metrics (Supplementary Table S4). Thus, for node-level analysis, we used the following landscape metrics: mean matrix resistance of the focal site and site isolation. For analyses at the link level, we used: mean matrix resistance and geographic distance between site pairs. Landscape metrics related to forest amount and matrix resistance were negatively correlated (r<−0.7; Supplementary Tables S4 and S5), and thus we used the matrix resistance as a proxy to discuss habitat loss and fragmentation.
Genetic variability and differentiation may reflect not only contemporary landscape structure but also historical processes such as past gene flow and population fluctuations (Zellmer and Knowles, 2009). Thus, along with contemporary landscape metrics, we also calculated historical effective population size in each site (metric for node-level analyses) and historical genetic connectivity among sites (metric for link-level analyses), because of their potential effect on current genetic diversity and genetic differentiation. The historical effective population size (Ne) was calculated using coalescent analysis under the isolation with migration model (θ=4 μNe, coalescent or mutation parameter for a diploid genome). We assessed historical connectivity among populations estimating the migration parameter (M=4Nem /θ) to decouple the effects of historical and current connectivity in genetic differentiation. Finally, we also estimated the exponential growth parameter g (θt=θnow exp(−gt), where t is the time to coalescence in mutational units) to detect historical reduction in effective population size. The analyses were performed using a Markov chain Monte Carlo approach implemented in the Lamarc 2.1.9 software (Kuhner, 2006). The analyses were run with 10 initial chains of 10 000 and two final chains of 100 000 steps; the chains were sampled every 100 steps following 10 000 steps burn-in. We used the default settings for the initial estimate of θ. We ran the analyses three times to assess convergence and validate the results. Then, we generated combined results using Tracer v.1.4.1 and considered the results only when effective sample size was ⩾200.
Landscape structure effects on genetic variables
We modeled the genetic response variables in relation to explanatory covariates using generalized linear mixed model. Models were defined according to the genetic response variables (see Figure 2 for expected relationship between genetic response variables and explanatory covariates) as node level (estimated for each site: He, AR and FIS) or link-level analyses (between sites within landscapes: FST, GST' and Jost's D).

Predicted effects of landscape structure on genetic response variables at the node and link levels of E. edulis. Genetic parameters are on the y axis and landscape metrics are on the x axis. For the node level, the black continuous line represents He and AR, and the dotted line represents the FIS. For the link level, the dotted line represents the pairwise genetic differentiation (FST, GST' and Jost's D).
To account for the presence of autocorrelation at node-level analyses, we tested models with distinct spatial covariance structures (Gaussian, exponential and spherical) using the restricted maximum-likelihood method in a generalized linear mixed model and compared with a model without spatial covariance structure. These models contained all explanatory covariates as fixed effects and landscapes and sites as random effects, to account for landscape non-independence and pseudoreplication of sites within landscape. To find the best spatial covariance structure, we compared the models using Akaike information criteria (AIC—see the explanation below). Using the best spatial covariance structure and landscape and sites as random effect, we finally tested a set of models using the package ‘nmle’ (Pinheiro et al., 2014) in the R software (R Development Core Team, 2013).
For the analyses at the link level, it is expected that the data within the same landscape are not independent (pairwise differentiation and landscape metrics), which would require a Mantel and Partial Mantel test. However, owing to the many cases of missing data between landscapes, we performed a generalized linear mixed model using landscapes as random effect to account for non-independence between landscapes. We also used the maximum-likelihood population-effects parameterization, in which the covariate structure is fit for the specific dependence between values in a matrix, to account for pairwise data non-independence (Clarke et al., 2002). The models for link level were fitted using SAS PROC MIXED (SAS University Edition) and the covariance structure was coded through toeplitz(1) (Selkoe et al., 2010). All models (for link and node levels) were fitted using the maximum-likelihood method, which is the best for assessing the influence of fixed effects, for a given random structure (Bolker et al., 2009).
For the node and link levels, we built a set of nested models that comprised all combinations of one to two explanatory covariates. We used a maximum of two variables in our models because of the limited sample size (n=16 for the node and n=19 for the link levels). We also built a null model without covariates to compete with the set of nested models. All models had normally distributed residuals, which we confirmed using the Shapiro–Wilk normality test.
We calculated AIC corrected for small sample sizes (AICc) and the difference of each model and the best model: ΔAICci (where i represents each model). We also estimated Akaike’s weight of evidence (wAICc) as the relative contribution of model i to explain the observed pattern, given a set of competing models (Burnhan and Anderson, 2002). Models with ΔAICc<2 or wAICc>0.1 were considered as equally plausible to explain the observed pattern (Zuur et al., 2009). The AIC-based analyses and the coefficient plots of the best-fitted models were obtained using R software (R Development Core Team, 2013). The coefficient plots of the best-fitted models were calculated to verify the significance of coefficient values associated with each explanatory covariate.
Results
Genetic variability, differentiation and demography history
All pairs of microsatellite loci were in linkage equilibrium (all P>0.05) and there was no evidence of genotyping errors or null alleles (results not shown). All loci presented high genetic variability (Supplementary Information and Supplementary Table S6), but the observed heterozygosity differed from the expectation under Hardy–Weinberg equilibrium for all loci (all P<0.001).
He and AR were high in all populations ranging from 0.716 to 0.864 and 6.2 to 9.2, respectively (Table 1). FIS was also high and significant for all populations, ranging from 0.101 to 0.295; and the FIS accounting for the potential impacts of null alleles ranged from 0.024 to 0.148 (Table 1). Populations were significantly differentiated (FST=0.116, s.e.=0.012, P<0.001, GST'=0.524, 95% confidence interval=0.498–0.548, Jost's D=0.461, 95% confidence interval=0.437–0.484) with pairwise FST ranging from 0.002 to 0.200, GST' ranging from 0.03 to 1 and Jost's D ranging from 0.026 to 1 across landscapes (Supplementary Information and Supplementary Table S7). Slatkin's RST (RST=0.077, s.e.=0.048) was not significantly different from FST (P=0.852), thus we followed the analyses using the FST parameter. The coalescence analysis showed that historical population sizes were constant in all sites (Table 1), as the confidence intervals of the exponential growth parameter include zero. It also showed high historical genetic connectivity between sites within landscapes (Supplementary Information and Supplementary Table S10), supporting the assumption that genetic diversity was relatively homogeneously distributed within landscapes before fragmentation.
Landscape structure effects on genetic variables
Node-level analyses: genetic variability
For the node-level analyses, ΔAICc and wAICc indicated that models without a spatial covariance structure were considerably better for modeling He, AR and FIS than those with spatial covariance structures (Gaussian, exponential and spherical) (Supplementary Information and Supplementary Table S11).
He was best explained by the model that contained historical effective population size alone (wAICc=0.83; Table 2), with sites with higher historical effective population size showing higher genetic diversity (Figure 3a). The slope coefficient of historical effective population size was positive and statistically significant (see Supplementary Figure S2 for coefficient plot).

Relationship of He and historical effective population size (a), AR and historical effective population size (b), AR and matrix resistance (c) and pairwise FST and matrix resistance (d) for 16 sites of E. edulis in Atlantic forest remnants of São Paulo state, Southeast Brazil. Symbols represent the five landscapes: Caetetus (closed triangle), Carlos Botelho (closed square), Corumbatai (closed circle), Dinamérica (closed inverted triangle) and Tambaú (closed lozenge).
AR was best explained by a model that combined historical effective population size and matrix resistance (wAICc=0.65; Table 2). The slope coefficients were statistically significant for both explanatory covariates (see also Supplementary Information and Supplementary Figure S3 for coefficient plot), but historical effective population size had stronger effects on AR. The AR was higher in sites with higher historical effective population size and lower matrix resistance (Figures 3b and c). Although sites with high matrix resistance showed enormous variation in AR, sites that were inside landscapes with high percentage of forest cover presented similar results. The model with historical effective population size alone also explained the variation observed in AR (wAICc=0.31), presenting a statistically significant slope coefficient (see also Supplementary Information and Supplementary Figure S4 for coefficient plots). The variation observed in inbreeding coefficients (FIS) was not explained by any of our competing models (Table 2), with the null model being plausible to explain the observed patterns (ΔAICc <2 and wAICc >0.1).
Link-level analyses: genetic differentiation
Results of the models containing FST, GST' and Jost's D were consistently similar, that is, explanatory covariates contributed similarly to the models. Matrix resistance was the most influential factor, as it was the only explanatory covariate that appeared in all best models (Table 3). Matrix resistance was important when it was the only factor in the models (wAIC for FST=0.57, GST'=0.68 and Jost's D=0.68), but was also important in models with additive effects of historical migration (Nem, wAIC for FST=0.31, GST'=0.18 and Jost's D=0.18) and geographic distance (wAIC for FST=0.12, GST'=0.14 and Jost's D=0.14). The models containing matrix resistance, together with historical migration and geographic distance, and matrix resistance alone, had cumulative wAICc of 100% to explain all genetic differentiation measures (Table 3). Historical migration (Nem) and geographic distance, however, did not present statistically significant coefficients (see Supplementary Information and Supplementary Figure S5). The resistant matrices presented positive relationship with genetic differentiation (Figure 3d, GST' and Jost's D results are in Supplementary Information and Supplementary Figures S14 and S15, as they are very similar to FST results) and a statistically significant slope coefficient (see Supplementary Information and Supplementary Figures S5 and S13). Indeed, landscapes with high forest cover had similar genetic differentiation values; on the other hand, landscapes with little forest cover showed variation in this genetic response.
Discussion
Our hypothesis that inbreeding and genetic differentiation would be higher and genetic variability lower in more disturbed landscapes (lower forest amount, higher patch isolation and higher matrix resistance) was partially corroborated. As expected, we found that current landscape structure influences genetic differentiation, with matrix resistance being the most important predictor. The high historical migration between sites within landscapes showed that populations of E. edulis shared a common evolutionary history until very recently, and the genetic differentiation is most likely because of recent changes caused by habitat loss and fragmentation. In fact, the number of migrants per generation between pairs of populations was correlated neither to any landscape covariate nor to any measure of genetic differentiation, reinforcing that the genetic differentiation detected here is most likely due to present landscape structure. Genetic variability (heterozygosity and AR), however, was more strongly determined by historical effective population size. This indicates that genetic variability is more strongly influenced by the legacy left by populations in the past than by contemporary processes occurring at the landscape scale. Overall, our findings corroborate the hypothesis that fragmentation and habitat loss are negatively affecting genetic differentiation of seedlings while the genetic variability of seedlings is less affected.
Genetic variability
Despite the long anthropogenic disturbance and fragmentation of the Atlantic forest, our results show that E. edulis still presents high levels of genetic diversity in all analyzed populations. High genetic diversity and AR has been reported for E. edulis in other regions (Gaiotto et al., 2003; Conte et al., 2008).
The lack of consistent association between habitat loss and fragmentation and genetic diversity may be because of the relatively recent fragmentation of the Atlantic forest relative to the species life cycle (Collevatti et al., 2001; Kramer et al., 2008) and also high historical effective population size of this species (Galetti et al., 2013). Historical effective population size may have a strong influence on genetic diversity (Frankham, 1995), as this covariate has a high probability of best explaining the observed patterns in expected heterozygosity. The effects on genetic diversity of past population demography rather than contemporary processes occurring at the landscape scale has been observed in other organisms. In alpine butterfly, for example, genetic diversity was related to historical processes rather than contemporary forest cover (Keyghobadi et al., 2005b). In addition, the assessment of genetic variability using highly variable microsatellite loci may mask the reduction in heterozygosity because fragmentation can take longer to affect heterozygosity (Collevatti et al., 2001). Moreover, it is possible that habitat loss and fragmentation have not yet reduced the population size to a point that markedly changes the processes maintaining genetic diversity (Kramer et al., 2008). E. edulis occur in swampy areas and in high densities in fragment remnants (Matos et al., 1999). Indeed, we observed historical constant population sizes for all sites.
Inbreeding was important in shaping the genetic variability in seedlings of E. edulis. Despite the outcrossed mating system (tm=0.94, Gaiotto et al., 2003) and high genetic diversity, we found moderate inbreeding coefficients in all populations. For self-compatible species, outcrossing rate may be highly variable among populations because of differences in pollinator availability and flowering phenology (Barrett, 2002). Therefore, it is most likely that high frequency of mating between closely related individuals occurs in certain landscapes, as observed for other Neotropical tree species pollinated by animals (e.g., Caryocar brasiliense; Collevatti et al., 2001). Moreover, E. edulis is pollinated mainly by small-sized bees, which present relatively short flight distance (<840 m; Zurbuchen et al., 2010). Flight distance in small bees can be reduced by habitat fragmentation and isolation, even at short isolation distances (Zurbuchen et al., 2010), increasing inbreeding within fragments. However, inbreeding was not explained by historical effective population size or by any landscape metrics considered in this study.
Notwithstanding, the AR was affected by habitat loss and fragmentation. The different pattern in the effect of landscape features may be because of a higher temporal lag in genetic diversity and inbreeding in comparison with AR (Keyghobadi et al., 2005a). AR tends to respond faster than genetic diversity, because of the loss of rare alleles due to marked reduction in habitat (Young et al., 1996; Keyghobadi et al., 2005a). AR was influenced by historical effective population size and matrix resistance. This result shows that, although AR is strongly influenced by historical factors (historical effective population size), habitat loss, fragmentation and matrix type may also have an important role in shaping this genetic variable. Defaunation due to forest fragmentation also caused phenotypic changes in seed size of E. edulis in the same region, affecting seed dispersal (Galetti et al., 2013). Thus, allele loss may be a result of random genetic drift as well as limited gene flow due to reduction of abundance and mobility of pollinators and seed dispersers.
Genetic differentiation
Differently from genetic diversity, the genetic differentiation between sites within landscapes is most likely due to recent changes caused by habitat loss and fragmentation. Some organisms showed the same patterns as found in our study (Keyghobadi et al., 2005b; Zellmer and Knowles, 2009), whereas others showed the opposite (Orsini et al., 2008). These results highlight the importance of accounting for historical patterns, such as past landscape structure and historical demography, when analyzing the effect of contemporary landscape structure on genetic diversity and differentiation.
Landscape structure affected genetic differentiation (FST, GST' and Jost' D) between sites within landscapes. Pairs of sites with high matrix resistance were more highly differentiated. The coalescent analysis showed that the high genetic differentiation between sites within landscapes may be a result of ongoing fragmentation effects since these sites were historically connected (high historical number of migrants per generation) and had historical constant population growth. Our findings may be the result of disruption of gene flow due to lower mobility of the resilient pollinators and seed dispersers of E. edulis, reduction of current effective size and the decline in genetic variability (AR) because of fragmentation and habitat loss. These may cause a disruption of the migration–drift equilibrium, as alleles lost by random genetic drift may not be rescued by gene flow.
As commented above, results published elsewhere show important effects of defaunation owing to fragmentation in E. edulis seed size and, consequently, in seed dispersal (Galetti et al., 2013). In addition, E. edulis’ pollinators may be strongly affected by fragmentation and habitat isolation, which here are partially represented by matrix resistance. Matrix resistance has been pinpointed as an important factor affecting the mobility of individuals and also abundance, occurrence and genetic variability (Eycott et al., 2012; Lange et al., 2012). The change in forest cover may also change the abundance and richness of seed dispersers and pollinators (e.g., González-Varo et al., 2009; Martensen et al., 2012). Most fragments in the studied region of the Atlantic forest are too small (<50 ha; Ribeiro et al., 2009) to support populations of large forest-dwelling frugivorous birds that can move over large distances, such as toucans and cotingas (Galetti et al., 2013). These animals are important for long-distance dispersal (Nathan, 2006; Holbrook, 2011), and their loss can limit the gene exchange among populations, together with random genetic drift, leading to an increase in genetic differentiation among populations. Although others studies also found habitat loss effects on genetic structure of seedlings (e.g., Kattle et al., 2007), as far as we know this is the first study that reports the effects of matrix resistance on genetic differentiation of seedlings across multiple landscapes.
Although isolation by distance (i.e. geographic distance) is historically the most used hypothesis to explain genetic differentiation, geographic distance between pairs of sites within landscapes did not explain the variation in pairwise FST, GST' and Jost' D in this study. This means that matrix resistance is more biologically meaningful to explain genetic differentiation in seedlings, at least for forest-based ecological processes. In fact, given the scale of our analyses, Euclidean distance may be less important than matrix resistance in explaining genetic diversity and differentiation (see Lange et al., 2012 for similar results).
Conclusion
In conclusion, our results showed that even the human-modified Atlantic forest can harbor high genetic diversity. It is evident, however, that matrix quality and habitat loss determine genetic differentiation of a palm seedlings. Habitat loss and fragmentation effects on genetic diversity and inbreeding (He and FIS) are less important, or harder to detect, compared with those effects on AR and genetic differentiation (AR and FST, GST' and Jost' D). Others studies also show correlation between genetic differentiation and contemporary landscape features, and between genetic diversity and historical factors (i.e. Keyghobadi et al., 2005a). Matrix resistance was the most important factor explaining variation in AR and genetic differentiation (AR and FST, GST' and Jost' D). Our results highlight the importance of including landscape variables along with geographical distance in landscape genetics studies, such as matrix resistance, when seeking to understand the key ecological processes associated to dispersal, as these variables can shape microevolutionary processes. Moreover, our results give a warning about reduced genetic variability and increased genetic differentiation in seedlings due to habitat loss and fragmentation. As immediate restoration actions, the maintenance of landscapes with forest cover >50% (Tambosi et al., 2014) and permeable matrices to mobility of pollinators and frugivores may be an effective strategy to maintain microevolutionary processes.
Data archiving
Sampling locations (Supplementary Table S1), resistance weight used for matrix resistance (Supplementary Table S2 and S3), Pearson's correlation of landscape metrics (Supplementary Table S4 and S5), genetic characterization of loci (Supplementary Table S6), matrix of pairwise genetic differentiation (Supplementary Table S7–S9), mapping and classification landscapes (Supplementary Figure S1), coefficient plots (Supplementary Figures S2–S13) and results of GST' and Jost' D (Supplementary Figure S14 and S15) uploaded as online Supplementary material. The data of the manuscript ‘Contemporary and historic factors influence differently genetic differentiation and diversity in a tropical palm’ are deposited at Dryad: http://dx.doi.org/10.5061/dryad.6c33h
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J . (2008). Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17: 5177–5188.
Balkenhol N, Pardini R, Cornelius C, Fernandes F, Sommer S . (2013). Landscape-level comparison of genetic diversity and differentiation in a small mammal inhabiting different fragmented landscapes of the Brazilian Atlantic Forest. Conserv Genet 14: 355–367.
Barrett SCH . (2002). The sexual evolution of plant sexual diversity. Nat Rev 3: 275–284.
Bijlsma R, Loeschcke V . (2012). Genetic erosion impedes adaptive responses to stressful environments. Evol Appl 5: 117–129.
Bolker BM, Brooks ME, Clark CJ, Geange SW, Poulsen JR, Stevens MH et al. (2009). Generalized linear mixed models: a practical guide for ecology and evolution. Trends Ecol Evol 24: 127–135.
Burnhan KP, Anderson DR . (2002) Model Selection and Multimodel Inference: An Information-Theoretic Approach. Springer: New York, NY, USA.
Carvalho CS . (2010). Estrutura espacial genética de uma população de palmito juçara (Euterpe edulis em um fragmento florestal defaunado. Monograph, Universidade Estadual Paulista 'Júlio de Mesquita Filho'.
Chybicki IJ, Burczyk J . (2009). Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100: 106–113.
Clarke RT, Rothery P, Raybould AF . (2002). Confidence limits for regression relationships between distance matrices: estimating gene flow with distance. J Agr Biol Environ St 7: 361–372.
Collevatti RG, Grattapaglia D, Hay JD . (2001). Population genetic structure of the endangered tropical tree species Caryocar brasiliense, based on variability at microsatellite loci. Mol Ecol 10: 349–356.
Conte R, Sedrez dos Reis M, Mantovani A, Vencovsky R . (2008). Genetic structure and mating system of Euterpe edulis Mart. populations: a comparative analysis using microsatellite and alloenzyme markers. J Hered 99: 476–482.
Côrtes MC, Uriarte M, Lemes MR, Gribel R, Kress J, Smouse PE et al. (2013). Low plant density enhances gene dispersal in the Amazonian understory herb Heliconia acuminata. Mol Ecol 22: 5716–5729.
Eycott AE, Stewart GB, Buyung-Ali LM, Bowler DE, Watts K, Pullin AS . (2012). A meta-analysis on the impact of different matrix structures on species movement rates. Landscape Ecol 27: 1263–1278.
Fahrig L . (2003). Effects of habitat fragmentation on biodiversity. Annu Rev Ecol Evol Syst 34: 487–515.
Frankham R . (1995). Relationship of genetic variation to population size in wildlife. Conserv Biol 10: 1500–1508.
Gaiotto FA, Brondani RPV, Grattapaglia D . (2001). Microsatellite markers for heart of palm—Euterpe edulis and E. oleracea Mart (Arecaceae). Mol Ecol Notes 1: 86–88.
Gaiotto FA, Grattapaglia D, Vencovsky R . (2003). Genetic structure, mating system, and long-distance gene flow in heart of palm (Euterpe edulis Mart.). J Hered 94: 399–406.
Galetti M, Fernandez JC . (1998). Palm heart harvesting in the Brazilian Atlantic forest: changes in industry structure and the illegal trade. J Appl Ecol 35: 294–301.
Galetti M, Guevara R, Côrtes MC, Fadini R, Von Matter S, Leite AB et al. (2013). Functional extinction of birds drives rapid evolutionary changes in seed size. Science 340: 1086–1090.
González-Varo JP, Arroyo J, Aparicio A . (2009). Effects of fragmentation on pollinator assemblage, pollen limitation and seed production of Mediterranean myrtle (Myrtus communis. Biol Conserv 142: 1058–1065.
Goudet J . (2002). FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.2). Available at: www2.unil.ch/popgen/softwares.fstat.htm (last accessed 10 June 2013) (updated from Goudet, 1995).
Hardy OJ, Charbonnel N, Fréville H, Heuertz M . (2003). Microsatellite allele sizes: a simple test to assess their Significance on genetic differentiation. Genetics 4: 1467–1482.
Hardy OJ, Vekemans X . (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Hedrick PW . (2005). A standardized genetic differentiation measure. Evolution 59: 1633–1638.
Holbrook KM . (2011). Home range and movement patterns of Toucans: implications for seed dispersal. Biotropica 43: 357–364.
Jost L . (2008). Gst and its relatives not measure differentiation. Mol Ecol 17: 4015–4026.
Kettle CJ, Hollingsworth PM, Jaffré T, Moran B, Ennos RA . (2007). Identifying the early genetic consequences of habitat degradation in a highly threatened tropical conifer, Araucaria nemorosa Laubenfels. Mol Ecol 16: 3591.
Keyghobadi N, Roland J, Matter SF, Strobeck C . (2005a). Among- and within-patch components of genetics diversity respond at different rates to habitat fragmentation: an empirical demonstration. Proc R Soc 272: 553–560.
Keyghobadi N, Roland J, Matter SF, Strobeck C . (2005b). Genetic differentiation and gene flow among populations of the alpine butterfly, Parnassius smintheus, vary with landscape connectivity. Mol Ecol 14: 1897–1909.
Kramer AT, Fant JB, Ashley MV . (2011). Influences of landscape and pollinators on population genetic structure: examples from three Penstemon (Plantaginaceae) species in the great basin. Am J Bot 98: 109–121.
Kramer AT, Ison JL, Ashley MV, Howe HF . (2008). The paradox of forest fragmentation genetics. Conserv Biol 22: 878–885.
Kuhner MK . (2006). Lamarc 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinform Appl Notes 22: 768–770.
Lange R, Diekotter T, Schiffmann LA, Wolters V, Durka W . (2012). Matrix quality and habitat configuration interactively determine functional connectivity in a widespread bush cricket at a small spatial scale. Landscape Ecol 27: 381–392.
Martensen AC, Ribeiro MC, Banks-Leite C, Prado PI, Metzger JP . (2012). Associations of forest cover, fragment area, and connectivity with neotropical understory bird species richness and abundance. Conserv Biol 26: 1100–1111.
Matos DMS, Freckleton RP, Watkinson AR . (1999). The role of density dependence in the population dynamics of a tropical palm. Ecology 80: 2635–2650.
Meirmans PG, Hedrick PW . (2011). Assessing population structure: FST and related measures. Mol Ecol Resour 11: 5–18.
Mousadik A, Petit RJ . (1996). High level of genetic differentiation for allelic richness among populations of the argan tree Argania spinosa (L.) skeels endemic to Morocco. Theoret Appl Genet 92: 832–839.
Mühlner S, Kormann U, Schmidt-Entling MH, Herzog F, Bailey D . (2010). Structural versus functional habitat connectivity measures to explain bird diversity in fragmented orchards. J Landscape Ecol 3: 52–63.
Nathan R . (2006). Long-distance dispersal of plants. Science 313: 786–788.
Nei M . (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Orsini L, Corander J, Alasentie A, Hanski I . (2008). Genetic spatial structure in a butterfly metapopulation correlates better with past than present demographic structure. Mol Ecol 17: 2629–2642.
Pinheiro J, R Development Core Team. (2014). nlme: Linear and nonlinear mixed effects models. R package version 3.1-118. Available at: http://CRAN.R-project.org/package=nlme (last accessed 20 November 2014)..
Pinto N, Keitt TH, Wainright M . (2012). LORACS: JAVA software for modeling landscape connectivity and matrix permeability. Ecography 35: 388–392.
R Development Core Team. (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. Available at: www.R-project.org/ (last accessed 20 November 2014)..
Reis MS, Fantini AC, Nodari RO, Reis A, Guerra MP, Mantovani A . (2000). Management and conservation of natural populations in Atlantic rainforest: the case study of palm heart (Euterpe edulis Martius). Biotropica 32: 894–902.
Ribeiro MC, Metzger JP, Martensen AC, Ponzoni FJ, Hirota MM . (2009). The Brazilian Atlantic Forest: How much is left, and how is the remaining forest distributed? Implications for conservation. Biol Conserv 142: 1141–1153.
Selkoe KA, Watson JR, White C, Horin TB, Iacchei M, Mitaral S et al. (2010). Taking the chaos out of genetic patchiness: seascape genetics reveals ecological and oceanographic drivers of genetic patterns in three temperate reef species. Mol Ecol 19: 3708–3726.
Storfer A, Murphy MA, Spear SF, Holderegger R, Waits LP . (2010). Landscape genetics: where are we now? Mol Ecol 19: 3496–3514.
Tambosi LR, Martensen AC, Ribeiro MC, Metzger JP . (2014). A framework to optimize biodiversity restoration efforts based on habitat amount and landscape connectivity. Restor Ecol 22: 169–177.
Van Geert A, Van Rossum F, Triest L . (2008). Genetic diversity in adult and seedling populations of Primula vulgaris in a fragmented agricultural landscape. Conserv Genet 9: 845–853.
Wagner HH, Fortin M . (2013). A conceptual framework for the spatial analysis of landscape genetic data. Conserv Genet 14: 253–261.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Winter D, R Development Core Team. (2014). mmod: Modern measures of population genetic differentiation. R package version 1.2.1. Available at: http://CRAN.R-project.org/package=mmod (last accessed 20 November 2014).
White GM, Boshier DH, Powell W . (2002). Increased pollen flow counteracts fragmentation in a tropical dry forest. An example from Swietenia humilis Zuccarini. Proc Natl Acad Sci USA 99: 2038–2042.
Young A, Boyle T, Brown T . (1996). The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11: 413–418.
Zellmer AJ, Knowles LL . (2009). Disentangling the effects of historic vs contemporary landscape structure on population genetic divergence. Mol Ecol 18: 3593–3602.
Zurbuchen A, Landert L, Klaiber J, Muller A, Hein S, Dorn S . (2010). Maximum foraging ranges in solitary bees: only few individual have the capability to cover long foraging distance. Biol Conserv 143: 669–676.
Zuur AF, Ieno EN, Walker N, Saveliev AA, Smith GM . (2009) Mixed Effects Models and Extensions in Ecology with R. Springer: New York, NY, USA.
Acknowledgements
This work was supported by the competitive grants PRONEX CNPq/FAPEG/AUX PESQ CH 007/2009, CNPq 477843/2011-5, BIOTA/FAPESP 2007/03392-6 and the network Rede Cerrado CNPq/PPBio (project no. 457406/2012-7) that we gratefully acknowledge. CS Carvalho received a CAPES, CNPq (project no. 401258/2012-2) and Fapesp (project no. 2014/01029-5) scholarship and MCC received a CNPq (project no. 401258/2012-2) scholarship. RGC, MCR and MG have been continuously supported by grants and scholarships from CNPq. We thank S Nazareth and S Hieda for field assistance, São Paulo Fundação Florestal for permission to sample in State Reserves and many farm owners for permission to sample in private properties. We also thank the two ornithologists, MA Pizo and COA Gussoni, and the entomologist R Sampaio. We acknowledge JAF Diniz-Filho, D Boscolo, P Jordano, C Garcia and the anonymous reviewers for useful comments on the manuscript, and F Martello for his help in editing the graphs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article

Cite this article
da Silva Carvalho, C., Ribeiro, M., Côrtes, M. et al. Contemporary and historic factors influence differently genetic differentiation and diversity in a tropical palm. Heredity 115, 216–224 (2015). https://doi.org/10.1038/hdy.2015.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.30
This article is cited by
-
Examining the spatiotemporal variation of genetic diversity and genetic rarity in the natural plant recolonization of human-altered areas
Conservation Genetics (2023)
-
Anthropogenic threats and habitat degradation challenge the conservation of palm genetic resources—an appraisal of current status, threats and look-ahead strategies
Biodiversity and Conservation (2023)
-
Genetic resilience of Atlantic forest trees to impacts of biome loss and fragmentation
European Journal of Forest Research (2023)
-
Patterns of genetic diversity and structure of a threatened palm species (Euterpe edulis Arecaceae) from the Brazilian Atlantic Forest
Heredity (2022)
-
Matrix dominance and landscape resistance affect genetic variability and differentiation of an Atlantic Forest pioneer tree
Landscape Ecology (2022)
