
Abstract
It is well established that reproductive isolation often arises from genome incompatibilities and that genes encoding reproductive traits are less prone to introgression. Hybrid zones of Mytilus trossulus and Mytilus edulis provide an intriguing model to assess reproductive isolation. Although gene flow is restricted in North America, introgression is pervasive in the Baltic. This study aimed at analyzing the shape of multilocus clines across the Baltic contact zone between M. edulis and M. trossulus to infer mechanisms of restriction to gene flow. We use maximum likelihood methods to construct the best fitting individual clines for five markers located on biparentally inherited autosomes and paternally and maternally inherited mitochondrial DNA (mtDNA). Strong cline shape differences among markers suggest that reproductive isolation arising from genome-wide incompatibilities is weak, and that these discrepancies possibly result from genetic drift, hybrid zone movement or marker-specific selection. However, the finding of a common cline center for M7 lysin (involved in fertilization) and paternally transmitted mtDNA (causing nuclear-mitochondrial incompatibilities in hybrids) suggest that these loci may play a role in incomplete reproductive isolation.
Similar content being viewed by others

Introduction
In cases where reproductive isolation between two closely related species is not complete, secondary contact leads to hybridization and often to stable hybrid zones (Arnold, 1997). In some cases differential introgression of loci across a contact zone is observed. For instance, genes encoding factors directly involved in reproductive isolation and/or environmental adaptation are often subject to adaptive selection, which leads to restricted gene flow and clinal allele distribution across hybrid zones. The strength of such selection is reflected in the slope of the allele frequency cline (Barton and Hewitt, 1985). In contrast, gene flow can be less restricted for those loci that do not decrease hybrid survival, reproductive performance or local adaptation, and are not closely linked to such loci.
Thus, distinct selection regimes can lead to differential introgression among different genes across the same hybrid zone. Where such discordances have been reported, this has been commonly attributed to natural selection (directly or indirectly through genetic hitchhiking) on at least one of the loci (McDonald, 1994). However, discordant differentiation patterns among genes can also result from factors others than selection (Bierne et al., 2003). For instance, random genetic drift introduces a large amount of stochastic variation in the evolution of unlinked neutral loci (Bierne et al., 2003). This heterogeneity may be inflated in secondary contact zones by the combined effect of hybridization and recombination. Sharp clinal variation can also be observed for neutral markers as a transitory stage immediately after secondary contact (Edwards and Skibinski, 1987). As even a large heterogeneity in patterns of genetic differentiation among different genetic markers is often consistent with genetic drift expectations (Bierne et al., 2003), selection can only be inferred if its effects exceed the expectations from these stochastic effects.
Hybrid zones of the marine mussels, Mytilus edulis and Mytilus trossulus represent an excellent model system to investigate mechanisms underlying gene flow between hybridizing taxa and to assess the evolutionary implications of hybridization. These species live in marine habitats mostly in allopatry but hybridize in several zones of contact (reviewed in Koehn, 1991). The outcome of hybridization between these Mytilus species can vary significantly, as exemplified by the hybrid zones in North America and Europe (reviewed in Riginos and Cunningham, 2005). Introgression is very restricted for both mitochondrial and nuclear loci between these two species in North American hybrid zones (Saavedra et al., 1996). In contrast, introgression is pervasive for nuclear DNA and mitochondrial DNA (mtDNA) in Baltic populations. The pattern of introgression is particularly noteworthy for mtDNA, given that mussels display two types of mtDNA genomes that are transmitted separately by males and females, in a mode of transmission termed doubly uniparental inheritance (Skibinski et al., 1994; Zouros et al., 1994): Females transmit the maternally inherited mtDNA genome to all offspring, whereas males transmit the paternally inherited mtDNA genome only to sons. Thus, males are usually heteroplasmic and females are usually homoplasmic. Maternally and paternally inherited mtDNA of Baltic M. trossulus has been almost completely replaced by introgressed M. edulis mtDNA molecules of a maternal origin (Quesada et al., 1999, 2003). However, European M. edulis and M. trossulus remain genetically distinct despite of extensive nuclear and mitochondrial introgression (Kijewski et al., 2006) and maintain their morphological (McDonald et al., 1991) and allozymic identity (Väinölä and Hvilsom, 1991).
The putative role of genes involved in hybrid incompatibilities or species-specific adaptations to explain these contrasting patterns of introgression among different genes remains controversial. It was suggested that Baltic M. trossulus might be considered as a hybrid swarm without reproductive barriers (Riginos and Cunningham, 2005). Under this scenario, the observed genetic differences are supposed to have arose mainly from local adaptation and/or genetic drift (Riginos and Cunningham, 2005). In contrast, several studies suggest that partial pre-zygotic and/or post-zygotic isolation may be operating in this hybrid zone (Väinölä and Hvilsom, 1991; Bierne et al., 2003). Among other mechanisms, assortative gamete interaction has been shown to cause pre-zygotic isolation in other Mytilus hybrid zones (Bierne et al., 2002).
It is interesting to note that pervasive gene flow between genetically distinct populations in the Baltic was not only observed in Mytilus, but also has been described in several occasions involving various taxa. The Baltic population of the clam Macoma balthica was found to be a genetic mixture between the Pacific M. b. balthica and the Atlantic M. b. rubra (Nikula et al., 2008). Furthermore, analysis of ancient DNA suggests that the recently extinct Baltic sturgeon population was of hybrid origin (Acipenser oxyrinchus × Acipenser sturio) (Tiedemann et al., 2007). As pervasive hybridization might be more widespread among Baltic animal populations of different taxa, it would be interesting to know whether there is a common underlying pattern.
The study presented here aims to analyse multilocus clines across the Baltic contact zone between M. edulis and M. trossulus to infer the mechanisms controlling gene flow across the Baltic Mytilus hybrid zone. We surveyed variation at three nuclear (ITS, M7 lysin, EFbis) and two mitochondrial (paternally and maternally transmitted mtDNA) loci. Internal transcribed spacer (ITS) markers have been extensively used as the diagnostic loci in Mytilus populations (for example, Riginos et al., 2002). M7 lysin is an acrosomal protein responsible for dissolving the egg vitelline envelope and having first polar body releasing function (Takagi et al., 1994). This protein is suspected to be a C-type lectin and exhibits carbohydrate-binding activity. Biochemical assays showed that carbohydrate recognition may play an important role in the gamete recognition process (Togo and Morisawa, 1997), and evidence exists for positive selection acting on the M7 lysin gene in sympatric and allopatric Mytilus populations (Riginos and McDonald, 2003; Riginos et al., 2006; Springer and Crespi, 2007). Similarly, the EFbis gene encodes an essential component of the translational apparatus and evidence has been recently provided for this locus being affected by a selective sweep in M. edulis populations (Faure et al., 2008). Finally, incompatibilities between nuclear- and mitochondrial-encoded factors as well as between paternally and maternally transmitted mitochondria have been invoked to explain reduced hybrid fitness in North American Mytilus hybrid zones (Saavedra et al., 1996).
We sample the Baltic hybrid zone and use the theoretical framework developed by Szymura and Barton (1986, 1991) for comparing the structure of clines for five individual loci. We assess the degree of reproductive isolation between the Baltic Mytilus populations. Furthermore, we evaluate whether the observed genetic patterns are compatible with purely neutral processes or point towards natural selection. There are mainly two alternative patterns, which can be expected from this investigation. First, all assessed loci show a widely concordant genetic structure and cline shape. This would imply that strong selective forces act on many loci indicating reduced hybrid fitness and restricted gene flow. This would affect even neutral markers because of genetic linkage. Alternatively, discordant genetic structure and cline shapes would indicate that different mechanisms such as locus-specific selection and/or stochastic forces control gene flow across this hybrid zone.
Materials and methods
Sampling sites and definition of transects
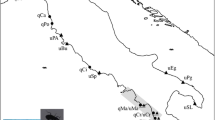
Mytilus specimens were collected along a transect from the following Baltic sites: Helgoland (Germany), Tjärnö (West coast of Sweden), Århus (Denmark), Kiel (Germany), Warnemünde (Germany), Hel (Poland) and Askö (East coast of Sweden) (Figure 1). A total of 11–20 animals per site were brought alive to the laboratory, dissected, frozen in liquid nitrogen and stored at −80 °C. Locality distances were measured along a line along the transect (dashed line in Figure 1), starting at its western edge in Helgoland (0 km), continuing over Tjärnö (678 km), Århus (1049 km), Kiel (1257 km), Warnemünde (1508 km) and Hel (1950 km), and ending in Askö (2450 km).

Location of the Mytilus populations analyzed in this study. The dashed line indicates how distances between locations were estimated.
Genotype assessment
The investigation of M7 lysin variability was performed by analyzing transcripts based on a reverse transcriptase-PCR (RT-PCR). Total RNA was extracted using Trizol reagent (Invitrogen, Karlsruhe, Germany) following the manufacturer's instructions using ∼200 mg of frozen tissue. Synthesis of cDNA was performed using the cDNA Synthesis Kit (Bioline GmbH, Luckenwalde, Germany). PCR primers (Mel-8: 5′-TATAAACCACGTCACGGGGG-3′; Mel-9: 5′-CCTTGTACGAATCGTCAGAT-3′) targeting a 414 bp fragment of the M7 lysin transcript were designed using earlier published nucleotide sequences (Riginos and McDonald, 2003). PCR was performed using 0.01 units GoTaq Flexi polymerase (Promega, Mannheim, Germany) with the following PCR conditions: 36 cycles with denaturation at 94 °C (for 20 s but for 5 min for the first cycle), annealing at 55 °C (for 20 s) and extension at 72 °C (for 45 s but 10 min for the final cycle). PCR conditions were slightly modified, when PCR fragments had to be cloned: 0.004 units Phusion High-Fidelity DNA Polymerase (Finnzymes, Espoo, Finland) were used and the PCR was performed using 30 cycles and an initial denaturation at 98 °C for 2 min.
For screening of alleles specific to M. edulis and M. trossulus, a RT-PCR-RFLP (restriction fragment length polymorphism) assay was developed based on M7 lysin nucleotide sequences from North American allopatric populations of M. edulis (Woods Hole, Atlantic coast) and M. trossulus (Penn Cove, Pacific coast) (Riginos and McDonald, 2003). RT-PCR products were cut with the restriction endonucleases TaqI (Fermentas, St. Leon Rot, Germany) and AluI (NEB, Frankfurt/M., Germany). The enzyme TaqI cleaves the M. trossulus M7 lysin PCR product in two fragments of 238 and 178 bp, whereas AluI cleaves the M. edulis M7 lysin PCR product in three fragments of 215, 136 and 63 bp. Further phylogenetic analysis (see Results) confirmed that M. edulis and M. trossulus M7 lysin alleles are clearly belonging to separate evolutionary lineages.
Further nuclear and mitochondrial markers were analyzed using DNA extracted after the cetyltrimethylammonium bromide (CTAB) procedure according to Skibinski et al. (1994). ITS alleles specific to M. edulis and M. trossulus were detected based on a PCR-RFLP approach according to Heath et al. (1995). As the ITS locus is a multicopy operon (Riginos et al., 2002), it was not analyzed for those parameters assuming a Mendelian inheritance (that is, Hardy–Weinberg equilibrium, cline analysis). The intron of the elongation factor 1 alpha (EFbis) was analyzed as an additional nuclear marker to assess alleles specific to M. edulis and M. trossulus according to Kijewski et al. (2006). This genomic region was PCR amplified using primers according to Bierne et al. (2003) that bind alleles of both species. Subsequently, PCR products were digested using the restriction endonucleases HhaI and RsaI in two separate reactions. Species-specific alleles were scored for each specimen based on the fact that HhaI cuts M. edulis specific alleles only and RsaI cuts M. trossulus specific alleles only. To analyse mtDNA variability, a mtDNA fragment of 527 bp encompassing the large subunit ribosomal RNA (lrRNA) was amplified with the universal primers 16S AR and 16S BR as used in Quesada et al. (2003). lrRNA PCR products were cut with the restriction endonucleases EcoRV, HaeIII and SpeI (all from NEB) according to Rawson and Hilbish (1995). Haplotypes were assessed according to Quesada et al. (2003) and pooled into three major categories: M. edulis F genome (FE-mitotype; maternally transmitted; haplotypes A, B), M. edulis M genome (ME-mitotype; paternally transmitted; haplotype C1) and masculinized mitochondrial genome (Mm-mitotype, paternally transmitted mitochondrial genome originating from a M. edulis F genome; haplotypes C and D). The distribution of an M. edulis M genome (ME-mitotype) was further assessed using specific primers targeting a fragment spanning parts of the ND2 and COIII genes as described by Skibinski et al. (1994) and Quesada et al. (2003). Phylogenetic analyses indicate that M. edulis and M. trossulus alleles/haplotypes of EFbis, ITS and mtDNA are clearly belonging to separate and highly divergent evolutionary lineages (Rawson and Hilbish, 1995; Riginos et al., 2002; Quesada et al., 2003; Kijewski et al., 2006; Faure et al., 2008).
Sequencing of M7 lysin RT-PCR products
To validate the new M7 lysin-RT-PCR-RFLP assay to characterize M. trossulus (T) and M. edulis (E) alleles, we cloned and sequenced M7 lysin RT-PCR products from mussels genotyped as T/T (n=3), E/E (n=6) and E/T (n=7). RT-PCR products were cloned using the TOPO TA Cloning Kit (Invitrogen). Cloned inserts were PCR amplified using vector primers and sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on a capillarity sequencer ABI PRISM 3100 (Applied Biosystems). As single PCR amplification may generate errors (for example, nucleotide substitutions, chimeric molecules) subsequently detected in single clones, haplotypes were determined using consensus sequences across cloned inserts. At least four independent clones were sequenced in case of homozygous individuals. At least three independent clones of each a priori classified allele were sequenced in case of heterozygous individuals. An allele was considered authentic when identical sequences were observed in at least two independent clones. The new sequences generated in this study are available from GenBank under accession numbers FJ649658–FJ649667.
DNA sequence data analysis
DNA sequences were multiple aligned using BIOEDIT (Hall, 1999) with reference to the M7 lysin sequences of allopatric populations of M. edulis (Woods Hole; AY 131165–131174), M. trossulus (Penn Cove; AY 131175–131182), and M. galloprovincialis (Samos; AY 131159–AY 131164) (Riginos and McDonald, 2003). An additional M7 lysin sequence was obtained from Takagi et al. (1994) and phylogenetically affiliated as M. galloprovincialis allele (Riginos and McDonald, 2003). Statistics describing nucleotide variation were calculated using DnaSP (Rozas et al., 2003). Nucleotide sequences were translated into amino acid sequences using BIOEDIT. Data were searched for evidence of recombination using the methods RDP, GENCONV, MaxChi, Bootscan, Chimaera and SIScan, as implemented in the computer program RDP3 (Martin et al., 2005).
The best-fit model of nucleotide substitution was selected using the log-likelihood ratio test in Modeltest 3.6 (Posada and Crandall, 1998). A maximum likelihood tree was generated with PAUP 4.0b10 (Swofford, 2001) under the HKY85 model, using heuristic maximum likelihood searches with tree bisection-reconnection branch swapping.
The molecular clock hypothesis was evaluated for the M7 lysin gene by comparing the likelihood of the maximum likelihood tree enforcing a molecular clock and the likelihood for the corresponding non-clock maximum likelihood tree with a log-likelihood ratio test using PAUP 4.0b10 (degrees of freedom equaling the number of sequences minus two).
Test for selection
The neutrality test of McDonald and Kreitman (1991) was used to determine whether the ratio of synonymous (Ds) to non-synonymous (Dn) divergence is the same as the ratio of synonymous (Ps) to non-synonymous (Pn) polymorphism comparing M7 lysin nucleotide sequences of M. edulis and M. trossulus. The α-index was used to quantify the proportion of divergence between species that is driven by adaptive selection, given as α=1–(DsPn/DnPs) (Eyre-Walker, 2006).
The ratio of non-synonymous to synonymous substitutions (KA/KS=ω) is a measure of the history and mode of selection acting on a gene or gene region. When ω<1, the gene is under selective constraint; when ω=1, the gene evolves without constraint on amino acid replacements; and when ω>1, there is evidence that positive selection has acted to promote amino acid replacements (Hughes and Nei, 1988). We estimated ω in all pairwise comparisons between M. edulis and M. trossulus sequences using the Jukes–Cantor correction for multiple hits as implemented in DnaSP. Variation of ω across the M7 lysin gene was visualized by a 50-bp sliding window plot. Where KA was greater than KS, the significance of this difference was tested following Zhang et al. (1997). To control the false discovery rate for multiple tests, P-values were adjusted using the sequential Bonferroni correction (Rice, 1989).
Sites under positive or negative selection across the M7 lysin gene were also investigated using a single likelihood ancestor counting analysis (Kosakovsky Pond and Frost, 2005a) as implemented in the DataMonkey server (Kosakovsky Pond and Frost, 2005b). This method uses a phylogenetic tree derived from a set of nucleotide sequences to estimate a global ω. Utilizing this gene tree, codon ancestral sequences are reconstructed using maximum likelihood. This allows to calculate codon-specific KA and KS values to analyse whether a site is under positive (KA>KS) or negative (KA<KS) selection.
Population structure and cline shape analysis
Deviation from Hardy–Weinberg equilibrium was tested with Arlequin software (Excoffier et al., 2005) and P-values were adjusted using Bonferroni correction (Sokal and Rohlf, 1995).
Cline shape was analyzed based on a method originally developed by Szymura and Barton (1986, 1991) in a maximum likelihood framework using the software ANALYSE (Barton and Baird, 1999). This methodological framework was used to relate geographic distance and allele frequencies to estimate different cline shape parameters from the West side of the cline (Helgoland) to the East side of the cline (Inner Baltic: Hel and Askö). The cline center (c) is the position where the estimated marker frequency equals to 0.5 and is given by the distance (km) from the West side of the cline (Helgoland). The cline width (w) is defined as 1/slope at the cline center. Among other parameters, the software allows estimating the marker frequencies P at the West (PWest) and the East (PEast) side of the cline, and the size of the central barrier to gene flow (B).
Each locus was analyzed separately to identify the best out of two models that describe cline shape with the fewest numbers of parameters. The two-parameter model estimates c and w only assuming that theta and B are one and zero, respectively, on both sides of the cline whereas the marker frequencies PWest and PEast were fixed at the observed allele frequencies. The four-parameter model estimates c, w as well as PWest and PEast at both sides of the cline. The ANALYSE software allows searching the parameter space using the Metropolis–Hastings algorithm giving a likelihood value for each parameter fit (lnL). Twenty independent runs employing 30 000 iterations were performed for each locus and each model to explore the parameter space using different starting points. The best fit model for each locus was determined by comparing likelihood values based on a log-likelihood ratio test: twice the difference between likelihood values was compared with a χ2-distribution and degrees of freedom equal to the number of independently estimated parameters. To estimate the 95% confidence interval of a particular parameter, the likelihood profile around the best estimate was explored by constraining the parameter of interest stepwise to values larger and smaller than the best estimate whereas all other parameters were free to vary (Phillips et al., 2004). The 95% support limit of a parameter was found within two lnL units (lnLmax−2).
We further compared how the M. edulis specific allele and mitotype frequencies decrease from West to East between pairs of loci considering the full transect using a non-parametric method suggested by Tsutakawa and Hewett (1977). This test compares the equality of the allele frequency at different geographical distances between two markers. We performed pairwise comparisons between markers to compare all clines with each other. First, the allele/haplotype frequencies of the two markers to be compared were plotted against geographical distance in the same graph. Second, a quadratic curve was fitted to the combined set of data points by weighting the least squares. Third, for each marker, we determined the number of data points above and below the fitted quadratic curve. Differences between markers in the number of data points above and below the fitted quadratic curve were tested using a Fisher's exact test. To correct for multiple tests, P-values were adjusted using the sequential Bonferroni correction (Rice, 1989).
Results
Genotyping of nuclear loci
Specimens were genotyped for the nuclear loci M7 lysin (67 in total), EFbis (111 in total) and ITS (122 in total). Alleles were phylogenetically classified as originating either from M. edulis (E) or M. trossulus (T) (Table 1). Frequencies of M. edulis specific alleles are depicted in Figure 2. No deviations from Hardy–Weinberg expectations were found for the nuclear markers EFbis and M7 lysin. Given that species-specific alleles/haplotypes of all markers used in this study are fixed in allopatric populations of North American M. trossulus, it is remarkable that in the Baltic no specimens were found that had only M. trossulus specific nuclear alleles at all loci under investigation. Furthermore, specimens that were heterozygous for both types of alleles at all loci, as expected for a F1 hybrid, were absent.

Mytilus edulis specific allele and haplotype frequencies plotted against geographical distance of the transect across the Baltic. (a) Frequencies of the M. edulis specific alleles of three nuclear markers (ITS, M7 lysin and EFbis). (b) Frequencies of the maternally transmitted M. edulis F genome (FE-mitotype) and the paternally transmitted M. edulis M genome (ME-mitotype). The ME-mitotype frequency equals the frequency of ME/FE heteroplasmic males as shown in Table 2. North American allopatric populations of M. trossulus are fixed for the T allele at all loci.
To validate the newly developed RT-PCR-RFLP assay for M7 lysin, 10 nucleotide sequences (372 bp long each) were used to generate a maximum likelihood tree (Figure 3). This analysis also included the nucleotide sequences from alleles found in allopatric populations of M. edulis, M. trossulus and M. galloprovincialis (Takagi et al., 1994; Riginos and McDonald, 2003). Although M. trossulus and M. galloprovincialis M7 lysin alleles form distinct clusters, the M. galloprovincialis cluster is nested into the clade of its sister species M. edulis. A detailed comparison of M. edulis and M. galloprovincialis was beyond the scope of our investigation. For the purpose of this study, it is important to note that M. edulis and M. trossulus M7 lysin alleles are clearly belonging to separate evolutionary lineages. We did not find evidence of recombination among the aligned sequences with any of the six methods assayed in this study.

Maximum likelihood tree of M7 lysin. The first letter indicates species affiliation (M. trossulus: T; M. edulis: E; M. galloprovinacialis: G) followed by a code identifying the corresponding sequence. The sequences obtained in this study are specified by a one letter code (a–j). Sequences taken from Riginos and McDonald (2003) are specified by an alphanumeric code as originally published. The sequence named Takagi is from Takagi et al. (1994). Bootstrap values higher than 50 are shown above the branches.
Tests of selection on Baltic M7 lysin alleles
A McDonald–Kreitman test comparing Baltic M. edulis and M. trossulus M7 lysin sequences showed a significant (P=0.028) excess of fixed non-synonymous substitutions between species, consistent with positive selection on the M7 lysin gene (eight fixed synonymous substitutions; 13 fixed non-synonymous substitutions; nine polymorphic synonymous substitutions; two polymorphic non-synonymous substitutions). The α-index indicated that up to 95% of nucleotide divergence between these two species could be driven by adaptive evolution. The overall ω-value was 0.324 in comparison between Baltic M. trossulus and M. edulis, indicating purifying selection. Our KA/KS sliding window analysis using the same set of sequences indicated a ω-value larger than one for a region at the 5′-end (data not shown). Although ω-values larger than one are principally indicative of positive selection acting on the respective regions, the difference between KA and KS was not statistically significant in any of our sliding-window partitions (test by Zhang et al., 1997; P>0.05). However, the single likelihood ancestor counting analysis found significant (P=0.007) evidence of purifying selection at one cysteine codon site. Substitution rates did not deviate significantly from a molecular clock (log-likelihood ratio test; P=0.067).
mtDNA analysis
Restriction digestion of mitochondrial lrRNA PCR products was performed for a total of 111 individuals (Table 2). Restriction patterns allowed a classification of mtDNA phylogenetic origin and their paternal or maternal mode of transmission following Quesada et al. (2003). A single maternally transmitted mitotype was found: M. edulis F genome (FE- mitotype). Furthermore, there were two distinct paternally transmitted mitotypes: M. edulis M genome (ME-mitotype) and masculinized mitochondrial genome (Mm-mitotype; paternally transmitted mitochondrial genome originating from M. edulis F genome). In addition, specimens were classified as homo- or heteroplasmic (Table 2). Females were found to be homoplasmic for the FE mitotype at all sampling sites. Heteroplasmy was found to be restricted to males (P<0.01), as expected from doubly uniparental inheritance of mtDNA in mussels. Differences among males were predominantly found in respect to the type of paternally transmitted mtDNA and the frequency of heteroplasmy. Males found in Helgoland, Tjärnö, Århus, Kiel and Warnemünde were all heteroplasmic for FE/ME-mitotypes except for one single male in Warnemünde, which had two types of FE-mitotypes differing in their restriction pattern (but no ME-mitotype). In contrast, such males with two FE-mitotypes or males heteroplasmic for FE/Mm-mitotypes were predominat in Hel and Askö, whereas FE/ME-heteroplasmic males were absent from Askö and only observed at low frequency in Hel. In addition, in Hel and Askö there was a relatively high frequency of males being homoplasmic for the FE-mitotype. As noted earlier (Quesada et al., 2003), it is likely that some of these homoplasmic males might be in fact heteroplasmic for a masculinized genome that has not diverged, or that has diverged very little from the FE-mitotype from which it arose, hence still sharing the same restriction profile. Distribution of FE- and ME-mitotypes across the transect is given in Figure 2.
Population structure and cline shape analysis
Parameters describing cline shape of species-specific nuclear alleles (EFbis, M7 lysin) and paternally transmitted mtDNA were estimated following Szymura and Barton (1986, 1991) as implemented in the software ANALYSE (Barton and Baird 1999). Parameters for ITS and maternally transmitted mtDNA were not estimated as these markers are almost fixed or fixed throughout the transect. The cline shape is best described by a two-parameter model estimating cline center and cline width (Table 3). Cline center and cline width for EFbis, M7 lysin and paternally transmitted mtDNA in males are given in Table 3. Different cline centers between EFbis (829 km from Helgoland) on one hand and the M7 lysin and paternally transmitted mtDNA (1546 and 1736 km from Helgoland) on the other hand, are indicated by non-overlapping confidence intervals. Evidently, a larger and spatially more dense sampling scheme along the assayed transect may have resulted in narrower confidence intervals. Nevertheless, we argue that our data already provide a reasonable reflection of the width and location of clines across the Baltic hybrid zone.
Apart from estimating confidence intervals, the null hypothesis that clines of M7 lysin, EFbis and paternally transmitted mtDNA have the same cline center was tested using a G-test (Table 4) (Brumfield et al., 2001). To define the null log-likelihood, log-likelihoods were calculated for a series of six different potential cline centers encompassing the range of observed values. For each marker, a log-likelihood was estimated using a Metropolis–Hastings search but fixing the cline center at each of the six potential values while the cline width was free to vary. The null log-likelihood was the maximum log-likelihood obtained by summing the log-likelihoods from all three markers at each potential cline center (lnLmax=−10.35; c=1546). If there are significant differences in cline position, this log-likelihood should be significantly worse than the overall log-likelihood calculated from an unconstrained search (−2.92). The double of the absolute difference between both log-likelihoods (14.86) follows a χ2-distribution with two degrees of freedom (number of loci –1). The null hypothesis that these three markers share the same cline center was rejected (P<0.001). Subsequent pairwise G-tests among loci (not shown) indicate that M7 lysin and paternally transmitted mtDNA share the same cline center (P>0.05) in an area close to Warnemünde, which is significantly (P<0.001) different from the cline center observed for EFbis at Kattegat, located somewhere between Tjärnö and Århus.
We used a similar approach to test for significant differences in cline width among the three markers, where cline width was fixed while cline center was free to vary (data not shown). The null hypothesis that all three markers have the same cline width was not rejected (χ2=1.12, d.f.=2).
The existence of different water currents on the Northern and the South-Eastern Baltic coast could influence the distribution of larvae. To test whether these differences could affect the estimation of cline fit parameters, all the above analyses were repeated for Southern Baltic populations only (that is, excluding the two samples from Tjärno and Askö). The newly estimated cline shape parameters and the subsequent G-tests were entirely consistent with the result obtained with the complete data set considering all seven sampling sites (results not shown).
As the markers ITS and maternally transmitted mtDNA were excluded from the cline shape analysis, a non-parametric test according to Tsutakawa and Hewett (1977) was employed to test for differences on the extent in which the frequency of the M. edulis allele/haplotype decreases from West to East between pairs of loci (Table 5). Significant differences exist after sequential Bonferroni correction between EFbis and the remaining four markers, and between M7 lysin, maternally transmitted mtDNA, and ITS. This result further supports the existence of differences in the extent of introgression among loci resulting from hybridization (introgressive hybridization; Arnold, 1997).
Discussion
The staggered genetic structure of the Baltic Mytilus hybrid zone
Using five markers located on biparentally inherited autosomes and maternally and paternally inherited mtDNA, our study provides no evidence for the existence of strong reproductive isolation between M. edulis and Baltic M. trossulus, as there is no indication of genome-wide incompatibilities among the two taxa. One source of evidence for this conclusion is the discordant allele/haplotype distributions among the five loci assayed and the observation of different levels of introgression among loci (Figure 2; Tables 4 and 5). As noted earlier (Quesada et al., 1999; Riginos et al., 2002; Kijewski et al., 2006), introgressive hybridization is particularly strong for maternally inherited mtDNA and ITS, leading to the complete or nearly complete replacement of native M. trossulus alleles/haplotypes by alien M. edulis alleles/haploypes. In contrast, the distribution of alleles and haplotypes of the remaining three markers (EFbis, paternally transmitted mtDNA, and M7 lysin) can be described as a cline. Sharper differences in allele/haplotype frequency between inner and outer Baltic were found for M7 lysin and paternally transmitted mtDNA than for EFbis (Figure 2). Although our cline width estimation might be partially influenced by the length of the intervals among sampling sites, the estimates for these three loci seem rather large (average w=659 km; Table 3) for an organism as Mytilus. This is because earlier studies combining oceanographic circulation models with genetic data of Mytilus hybrid dispersal (into pure zones) indicate dispersal distances of 30–64 km in the United Kingdom (Gilg and Hilbish, 2003). Cline width provides indeed the most direct information on reproductive isolation, as it measures the extent of gene flow at the center of the zone, where hybrids are more likely to occur. Apart from cline width, log-likelihood tests show that M7 lysin and paternally transmitted mtDNA share a common cline center, which is displaced approximately one cline width in respect to the cline center of EFbis (Tables 3 and 4). This again supports our theory of weak reproductive isolation between Baltic Mytilus species, as genome-wide incompatibilities (post-zygotic isolation) are expected to shape clines with a common cline center. An additional line of evidence for low level of reproductive isolation between M. edulis and Baltic M. trossulus is the lack of M. trossulus individuals displaying native alleles/haplotypes at all five markers. In this respect, genome-wide levels of introgressive hybridization are likely even higher than reported here, as diagnostic markers are expected to be under stronger deterministic forces opposing the movement of alleles across the hybrid zone than randomly chosen neutral loci (Brumfield et al., 2001).
Interestingly, Väinölä and Hvilsom (1991) described concordant clines for four diagnostic allozymes across a relatively narrow transition zone of 100 km along the Øresund region. Subsequent work has shown that these loci do not segregate independently because they belong to the same linkage group (Beaumont, 1994). Nonetheless, it remains possible that stochastic influences (genetic drift) probably interact with one or more deterministic forces specific to these diagnostic loci to produce concordant clines for these markers and in dissociating the position and width of these clines from others (Barton, 1993).
Evolutionary forces shaping the Baltic hybrid zone
At the time of the influential review of Barton and Hewitt (1985), it seemed that the majority of hybrid zones were characterized by the occurrence of concordant clines. However, since then there have been an increasing number of studies reporting discordant clines, and this may be a rather frequent phenomenon (Barton, 1993; Jaarola et al., 1997; Brumfield et al., 2001; Payseur et al., 2004). There is a range of possible explanations to account for the discordance of clines in a hybrid zone (Barton, 1993). Extensive asymmetric introgression of alleles/haplotypes far beyond the limits of the hybrid zone (that is, ITS and maternally inherited mtDNA) might be explained by neutral introgression, movement of the hybrid zone, a founder event or selective advantage. Asymmetric mtDNA introgression could also result from gender-specific differences in pre-zygotic and post-zygotic reproductive barriers (Bierne et al., 2002; Rawson et al., 2003). Alternatively, small cline shifts of about one cline width as those observed for M7 lysin and paternally inherited mtDNA with respect to EFbis, may be the result of selection against certain gene combinations (Barton, 1993), chromosomal rearrangements (that is, Robertsonian fusions; Fel-Clair et al., 1996) or genetic drift (Barton, 1983). Given these various mechanisms, future studies have to disentangle the exact process, which led to the genetic structure of the Baltic Mytilus hybrid zone. We argue that particular attention should be paid to investigate a possible movement of the hybrid zone and to study patterns of differential selection. These mechanisms are considered as not mutually exclusive, resulting both in a non-equilibrium situation.
Paleoclimatic data indicate that oceanographic features and the distribution of marine species in the surrounding area of the Baltic have experienced drastic changes for the last 2 million years, thus providing ample opportunity for the movement of the Mytilus hybrid zone. The ancestors of the current populations of Baltic M. trossulus started to invade the Atlantic Sea from the Pacific during the Pleistocene (1.8 million years ago) or Holocene (10 000 years ago), with subsequent secondary contact and hybridization with the resident populations of M. edulis (Riginos and Cunningham, 2005). However, the colonization of the Baltic area was only possible after the last deglaciation, ∼7500 years ago, which allowed establishing a connection between the Baltic and the North Sea (Donner, 1995). Salinity in the Baltic has probably decreased because of freshwater input in the past (Donner, 1995). Thus, the area of contact and hybridization between M. edulis and Baltic M. trossulus has most likely moved (even repeatedly) under the influence of the colonization process and/or the changing salinity conditions. This suggests that geographical shifts in the position of the hybrid zone may have moved many molecular markers eastwards (like ITS and maternally inherited mtDNA), but left others behind (M7 lysin, paternally inherited mtDNA and EFbis). Under this scenario, the small shift of one cline width for M7 lysin and paternally inherited mtDNA with respect to EFbis could just be because of drift.
Alternative explanations assuming differential selection are, however, possible. This conclusion is based on the observation of concordant cline center positions between the loci M7 lysin and the paternally inherited mtDNA. This result is remarkable for two reasons. First, M7 lysin plays an important role in the fertilization process (Togo and Morisawa, 1997). On the basis of McDonald–Kreitman test, our study corroborates earlier findings (Riginos and McDonald, 2003) indicating that this locus is under positive selection, and that the deviation from neutrality is in the direction of an excess of fixed amino acid replacements between M. edulis and M. trossulus. Second, the special mode of doubly uniparental inheritance is characterized by a coupling between sex and mtDNA inheritance, implying strong interactions between nuclear and mtDNA encoding factors (Saavedra et al., 1996). Interestingly, the sharp shift in haplotype frequencies observed for paternally transmitted mtDNA is not seen for maternally transmitted mtDNA, suggesting that drift, or distinctive compatibility constraints between nuclear genes and mitochondrial genomes, or between paternally and maternally transmitted mitochondrial genomes are responsible for these contrasting patterns of variation between different mtDNA lineages. Consistent with this, the paternally transmitted mtDNA also shows restricted gene flow between trans-Atlantic Mytilus populations (Riginos et al., 2004). Our study showed that mitochondrial lineages are particularly distinct between the inner and outer Baltic in males (Table 2). The presence of male biased heteroplasmy provides evidence for a correct function of doubly uniparental inheritance. Given the distinctiveness in respect to mitotypes found in males, doubly uniparental inheritance specific nuclear-mitochondrial and/or mitochondrial-mitochondrial interactions could exist in the inner and outer Baltic. Indeed, such incompatibilities seem to block interspecies gene flow in American mussels (Saavedra et al., 1996). Thus, epistatic interactions could explain the concordant cline positions between these two unlinked markers, dissociating their position from that displayed by other markers.
Given the evidence for involvement of M7 lysin and mtDNA in reproductive processes, our data suggest the existence of a weak semi-permeable barrier to gene flow between Baltic Mytilus species. Such a semi-permeable barrier could oppose the movement of alleles across the hybrid zone for a reduced number of genes and linked markers. It is possible that the higher levels of introgression in Baltic than in American populations of M. trossulus and M. edulis could result from differences in the age of secondary contact among the hybridizing populations from each continent, leading to differences in the outcome of local adaptation and the strength of reproductive isolation (Riginos and Cunningham, 2005). However, it will be difficult to precisely distinguish between the effect of selection, neutral processes and processes such as movement of the entire hybrid zone. This is additionally hampered by the lack of precise historic data about this hybrid zone. However, given that genes involved in gamete function are candidates for reproductive isolation, we hypothesize that these genes should display similar cline shapes and positions to those of M7 lysin and paternally inherited mtDNA. To further evaluate this hypothesis, selective antibody production as described by Stuckas et al. (2009) will be one way to directly screen for such factors and their genes and to use monoclonal antibodies for functional assays to show their role in the fertilization process and in reproductive isolation. Such further assessment will not only be relevant to understand the dynamics of hybridization in Baltic Mytilus species but might also contribute to a better understanding of pervasive hybridization events, that is, as observed in other Baltic species (that is, Macoma balthica and extinct Baltic sturgeon).
References
Arnold M (1997). Natural Hybridization and Evolution. Oxford University Press: Oxford, New York.
Barton NH (1983). Multilocus clines. Evolution 37: 415–426.
Barton NH, Hewitt GM (1985). Analysis of hybrid zones. Annu Rev Ecol Syst 16: 113–148.
Barton NH (1993). Why species and subspecies? Curr Biol 3: 797–799.
Barton NH, Baird SJE (1999). Analyse: software for the analysis of geographic variation and hybrid zones. Ver. 1.03 for Macintosh. Available via http://www.helios.bto.ed.ac.uk/research/institutes/evolution/software/Mac/Analyse/index.html.
Beaumont AR (1994). Linkage studies in Mytilus edulis, the mussel. Heredity 72: 557–562.
Bierne N, David P, Boudry P, Bonhomme F (2002). Assortative fertilization and selection at larval stage in the mussels Mytilus edulis and M. galloprovincialis. Evolution 56: 292–298.
Bierne N, Daguin C, Bonhomme F, David P, Borsa P (2003). Direct selection on allozymes is not required to explain heterogeneity among marker loci across a Mytilus hybrid zone. Mol Ecol 12: 2505–2510.
Brumfield RT, Jernigan RW, McDonald DB, Braun M (2001). Evolutionary implications of divergent clines in an avian (Manacus: Aves) hybrid zone. Evolution 55: 2070–2087.
Donner J (1995). The Quaternary History of Scandinavia. Cambridge University Press: Cambridge.
Edwards CA, Skibinski DOF (1987). Genetic variation of mitochondrial DNA in mussels (Mytilus edulis and Mytilus galloprovincialis) populations from South West England and South Wales. Mar Biol 94: 547–556.
Excoffier L, Laval G, Schneider S (2005). Arlequin ver 3.1: an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Eyre-Walker A (2006). The genomic rate of adaptive evolution. Trends Ecol Evol 21: 569–575.
Faure MF, David P, Bonhomme F, Bierne N (2008). Genetic hitchhiking in a subdivided population of Mytilus edulis. BMC Evol Biol 8: 164.
Fel-Clair F, Lenormand T, Catalan J, Grobert J, Orth A, Boursot P et al. (1996). Genomic incompatibilities in the hybrid zone between house mice in Denmark: evidence from steep and non-coincident chromosomal clines for Robertsonian fusions. Genet Res 67: 123–134.
Gilg MR, Hilbish TJ (2003). The geography of marine larval dispersal: coupling genetics with fine-scale physical oceanography. Ecology 84: 2989–2998.
Hall TA (1999). BioEdit: a user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41: 95–98.
Heath D, Rawson PD, Hilbish TJ (1995). PCR-based nuclear markers identify alien blue mussel (Mytilus spp.) genotypes on the west coast of Canada. Can J Fish Aquat Sci 52: 2621–2627.
Hughes AL, Nei M (1988). Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335: 167–170.
Jaarola M, Tegelström H, Fredga K (1997). A contact zone with noncoincident clines for the sex-specific markers in the field vole (Microtus agrestis). Evolution 51: 241–249.
Kijewski T, Zbawicka M, Väinölä R, Wenne R (2006). Introgression and mitochondrial heteroplasmy in the Baltic populations of mussels Mytilus trossulus and M. edulis. Mar Biol 149: 1371–1385.
Koehn KR (1991). The genetics and taxonomy of species in the genus Mytilus. Aquaculture 94: 125–145.
Kosakovsky Pond SL, Frost SDW (2005a). Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22: 1208–1222.
Kosakovsky Pond SL, Frost SDW (2005b). Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21: 2531–2533.
Martin DP, Willianson C, Posada D (2005). RDP2: recombination detection and analysis from sequence alignments. Bioinformatics 21: 260–262.
McDonald JH, Seed R, Koehn RK (1991). Allozymes and morphometric characters of three species of Mytilus in the Northern and Southern hemispheres. Mar Biol 111: 323–333.
McDonald JH, Kreitman M (1991). Adaptive protein evolution at the Adh locus in Drosophila. Nature 354: 114–116.
McDonald JH (1994). Detecting neutral selection by comparing geographic variation in protein and DNA polymorphisms. In: Golding B (ed). Non-neutral Evolution: Theories and Molecular Data. Chapman and Hall: New York. pp 88–100.
Nikula R, Strelkov P, Väinölä R (2008). A broad transition zone between an inner Baltic hybrid swarm and a pure North Sea subspecies of Macoma balthica (Mollusca, Bivalvia). Mol Ecol 17: 1505–1522.
Payseur BA, Krenz JG, Machman MW (2004). Differential patterns of introgression across the X chromosome in a hybriod zone between two species of house mice. Evolution 58: 2064–2078.
Phillips B, Baired SJE, Moritz C (2004). When vicars meet: a narrow contact zone between morphologically cryptic phylogeographic lineages of the rainforest skink, Carlia rubrigularis. Evolution 58: 1536–1548.
Posada D, Crandall KA (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14: 817–818.
Quesada H, Wenne R, Skibinski DOF (1999). Interspecies transfer of female mitochondrial DNA is coupled with role-reversals and departure from neutrality in the mussel Mytilus trossulus. Mol Biol Evol 16: 655–665.
Quesada H, Stuckas H, Skibinski DOF (2003). Heteroplasmy suggests paternal cotransmission of multiple genomes and pervasive reversion of maternally into paternally transmitted genomes of mussel (Mytilus) mitochondrial DNA. J Mol Evol 57 (Suppl 1): S138–S147.
Rawson PD, Hilbish TH (1995). Evolutionary relationships among the male and female mitochondrial DNA lineages in the Mytilus edulis species complex. Mol Biol Evol 12: 893–901.
Rawson PD, Slaughter C, yund PO (2003). Patterns of gamete incompatibility between the blue mussels Mytilus edulis and M. trossulus. Mar Biol 143: 317–325.
Rice WR (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Riginos C, Sukhdeo K, Cunningham CW (2002). Evidence for selection at multiple allozyme loci across a mussel hybrid zone. Mol Biol Evol 19: 347–351.
Riginos C, McDonald JH (2003). Positive selection on acrosomal sperm protein, M7 lysin, in the three species of the mussel genus Mytilus. Mol Biol Evol 20: 200–207.
Riginos C, Hickerson MJ, Henzler CM, Cunningham CW (2004). Differential patterns of male and female mtDNA exchange across the Atlantic ocean in the blue mussel, Mytilus edulis. Evolution 58: 2438–2451.
Riginos C, Cunningham CW (2005). Local adaptation and species segregation in two mussel (Mytilus edulis × Mytilus trossulus) hybrid zones. Mol Ecol 14: 381–400.
Riginos C, Wang D, Abrams JA (2006). Geographic variation and positive selection on M7 lysin, an acrosomal sperm protein in mussels (Mytilus spp). Mol Biol Evol 23: 1952–1965.
Roff DA, Bentzen P (1989). The statistical analysis of mitochondrial DNA polymorphisms: Chi square and the problem of small samples. Mol Biol Evol 6: 539–545.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003). DnaSP, DNA polymorphism analysis by the coalescent and other methods. Bioinformatics 19: 2496–2497.
Saavedra C, Stewart DT, Stanood RR, Zouros E (1996). Species-specific segregation of gender-associated mitochondrial DNA types in an area where two mussel species (Mytilus edulis and M. trossulus) hybridize. Genetics 143: 1359–1367.
Skibinski DOF, Gallagher C, Beynon CM (1994). Sex-limited mitochondrial DNA transmission in the marine mussel Mytilus edulis. Genetics 138: 801–809.
Sokal RR, Rohlf FJ (1995). Biometry, 3rd edn. Freeman and Company: New York.
Springer SA, Crespi BJ (2007). Adaptive gamete-recognition divergence in a hybridizing Mytilus population. Evolution 61: 772–783.
Stuckas H, Messerschmidt K, Putzler S, Baumann O, Schenk JA, Tiedemann R et al. (2009). Detection and characterization of gamete specific molecules in Mytilus edulis using selective antibody production. Mol Reprod Dev 76: 4–10.
Szymura JM, Barton NH (1986). Genetic analysis of a hybrid zone between the fire-bellied toads Bombina bombina and B. variegata, near Cracow in Southern Poland. Evolution 40: 1141–1159.
Szymura JM, Barton NH (1991). The genetic structure of the hybrid zone between the fire-bellied toads Bombina bombina and B. variegata: comparisons between transects and between loci. Evolution 45: 237–261.
Swofford DL (2001). PAUP* Phylogenetic Analysis Using Parsimony, version 4.0b10. Sinauer: Suderland.
Takagi T, Nakamura A, Deguchi R, Kyozuka K (1994). Isolation, characterization, and primary structure of three major proteins obtained from Mytilus edulis sperm. J Biochem 116: 598–605.
Tiedemann R, Moll K, Paulus KB, Scheer M, Williot P, Bartel R et al. (2007). Atlantic sturgeons (Acipenser sturio, Acipenser oxyrinchus): American females successful in Europe. Naturwissenschaften 94: 213–217.
Togo T, Morisawa M (1997). Aminopeptidase-like protease released from oocytes affects oocyte surfaces and suppresses the acrosome reaction in establishment of polyspermy block in oocytes of the mussel Mytilus edulis. Dev Biol 182: 219–227.
Tsutakawa RK, Hewett JE (1977). Quick test for comparing two populations with bivariate data. Biometrics 33: 215–219.
Väinölä R, Hvilsom MM (1991). Genetic divergence and a hybrid zone between Baltic and North Sea Mytilus populations (Mytilidae: Mollusca). Biol J Linn Soc 43: 127–148.
Zhang J, Kumar S, Nei M (1997). Small-sample tests of episodic adaptive evolution: a case of primate lysozyme. Mol Biol Evol 14: 1335–1338.
Zouros E, Ball AO, Saavedra C, Freemann KR (1994). An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc Natl Acad Sci USA 91: 7463–7467.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft for financial support (TI 349/3-1). We are grateful to Martin Wahl and Ute Kossak for access to samples and to Katja Moll for technical assistance. We thank three anonymous referees for suggestions to improve the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stuckas, H., Stoof, K., Quesada, H. et al. Evolutionary implications of discordant clines across the Baltic Mytilus hybrid zone (Mytilus edulis and Mytilus trossulus). Heredity 103, 146–156 (2009). https://doi.org/10.1038/hdy.2009.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2009.37
Keywords
This article is cited by
-
Spatio-temporal variation in stable isotope and elemental composition of key-species reflect environmental changes in the Baltic Sea
Biogeochemistry (2022)
-
First description of a widespread Mytilus trossulus-derived bivalve transmissible cancer lineage in M. trossulus itself
Scientific Reports (2021)
-
The effects of cold stress on Mytilus species in the natural environment
Cell Stress and Chaperones (2020)
-
Combined genotyping, microbial diversity and metabolite profiling studies on farmed Mytilus spp. from Kiel Fjord
Scientific Reports (2018)
-
Temporal change of the Baltic Sea–North Sea blue mussel hybrid zone over two decades
Marine Biology (2017)
