
Abstract
Purpose
To test the hypothesis that undiagnosed patients with Fabry disease exist among patients affected by common heart disease.
Methods
Globotriaosylceramide in random whole urine using tandem mass spectroscopy, α-galactosidase A activity in dried blood spots, and next-generation sequencing of pooled or individual genomic DNA samples supplemented by Sanger sequencing.
Results
We tested 2,256 consecutive patients: 852 women (median age 65 years (19–95)) and 1,404 men (median age 65 years (21–92)). The primary diagnoses were coronary artery disease (n = 994), arrhythmia (n = 607), cardiomyopathy (n = 138), and valvular disease (n = 568). Urinary globotriaosylceramide was elevated in 15% of patients and 15 males had low α-galactosidase A activity. GLA variants found included R118C (n = 2), D83N, and D313Y (n = 7); IVS6-22 C>T, IVS4-16 A>G, IVS2+990C>A, 5′UTR-10 C>T (n = 4), IVS1-581 C>T, IVS1-1238 G>A, 5′UTR-30 G>A, IVS2+590C>T, IVS0-12 G>A, IVS4+68A>G, IVS0-10 C>T, IVS2-81–77delCAGCC, IVS2-77delC. Although the pathogenicity of several of these missense mutations and complex intronic haplotypes has been controversial, none of the patients screened in this study were diagnosed definitively with Fabry disease.
Conclusion
This population of patients with common heart disease did not contain a substantial number of patients with undiagnosed Fabry disease. GLA gene sequencing is superior to urinary globotriaosylceramide or α-galactosidase A activity in the screening for Fabry disease.
Similar content being viewed by others


Introduction
Fabry disease is an X-linked genetic disorder (OMIM 301500). The incidence of the disease has been estimated to be 1 in 117,000 live male births;1 however, recent newborn screening surveys suggest that the incidence may be 10 times higher.2,3,4 The most common complication of Fabry disease is cardiac dysfunction, which may include cardiomyopathy, atrio-ventricular conduction defects, arrhythmia, valvular dysfunction, and non-atherosclerotic coronary vascular disease.5,6 Increased stroke risk,7 progressive renal failure,8 and painful small-fiber neuropathy9 occur commonly as well.10 Fabry disease is caused by a deficiency of the lysosomal enzyme α-galactosidase A and accumulation of the glycosphingolipid globotriaosylceramide (Gb3) in most cells and organs, as well as an increase of Gb3 in urine.11,12,13 Urinary Gb3 is not found primarily in the filtrate, but mostly in shed renal tubular cells.13,14 Increased blood and urine Gb3 and globotriaosylsphingosine (lyso-Gb3) are considered important specific biomarkers in this disease, and are often used in screening and diagnosis for this disorder.15,16
Since cardiac manifestations of Fabry disease are common and largely non-specific in their clinical presentation, we screened for this disorder in patients with common heart disease. We hypothesized that there are patients with undiagnosed Fabry disease, particularly with later-onset mutations, among patients with all forms of heart disease seen in the general population, although the incidence may be higher in more specific types of heart disease, such as hypertrophic cardiomyopathy.17,18,19
Materials and methods
Patients
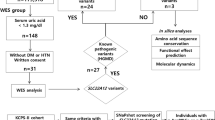
We screened for Fabry disease in a population of patients with multiple forms of cardiovascular disease (ClinicalTrials.gov identifier: NCT01019629). These included coronary artery disease, conduction or rhythm abnormalities, non-ischemic cardiomyopathy, and valvular dysfunction. The patients were ambulatory, had to be over 18 years of age, and were seen at a number of institutions in Dallas: Baylor Heart and Vascular Hospital, the Heart Hospital at Baylor Plano, Soltero Cardiovascular Research Center, and cardiology outpatient clinics. More than 95% of patients who were asked to participate in the study accepted and gave written informed consent. The institutional review board of the Baylor Research Institute provided oversight for the study. We estimated that about 0.5–2% of all patients screened would be positive for Fabry markers and the screening of at least 1,000 patients was considered adequate.
Screening for Fabry disease was performed by measuring urinary Gb3 in randomly collected samples of whole urine using ultra high pressure chromatography–tandem mass spectrometry, measuring α-galactosidase A activity in dried blood spots by flow injection analysis–tandem mass spectrometry, and testing for GLA gene mutations by parallel sequencing of the whole gene in pooled genomic DNA samples.
Urinary Gb3 analysis by mass spectrometry
Analysis was performed as previously described.20
α-Galactosidase A activity evaluation by tandem mass spectrometry
Enzyme analysis was performed as previously described.20
GLA gene analysis
GLA gene variants were searched for in pooled DNA using massively parallel sequencing21 or in individual genomic DNA samples using the Illumina MiSeq platform (Illumina, Inc.; San Diego, CA). Briefly, the SmartChip MyDesign TE system (WaferGen Bio-systems, Inc., Fremont, CA) was designed to enrich multiple samples for multiple resequencing targets simultaneously using a 4–polymerase chain reaction primer amplification strategy. The system consisted of SmartChip MyDesign chips that contain 5,184 nanowells. The separate, 100-nL reactions were then amplified using the Techne Prime Thermal Cycler (Techne, Staffordshire, UK), which was preprogrammed with the recommended thermal cycling program and equipped with an adapter plate for cycling 1 or 2 chips. After the polymerase chain reactions, pooled amplicons were collected from the chip(s) via centrifugation using single-use components supplied in the SmartChip TE Collection Kit. The library was then loaded on the MiSeq platform. Raw image files were processed by base-calling software with default parameters and the sequences of each individual were generated as 250 base pairs paired-end reads. Conventional Sanger sequencing was used to further analyze individual samples from selected patient DNA pools.
Results
A total of 2,256 consecutive patients were screened for Fabry disease; 1,404 were male (62.2%) and 852 were female (37.8%). The median age of women was 65 years (19–95) for women and men was 65 years (21–92) for men. Eighty-four patients were Hispanic, 124 were African Americans, and 2,048 were non-Hispanic Caucasians. All types of complications related to heart disease were represented in this study. Overall, the primary diagnosis was coronary artery disease in 994 patients, arrhythmia in 607, cardiomyopathy in 138, and valvular disease in 568. Patients often had more than one type of clinical cardiac abnormality (Figure 1). Table 1 describes physiologic and biochemical characteristics of the screened patient population with heart disease, including urine Gb3 and whole-blood α-galactosidase A activity. Table 2 describes the GLA gene variants that were identified. α-Galactosidase A was abnormally low in 1% (15/1,404) of men and 0.8% (7/852) of women (Figure 2). Urine Gb3 was abnormally elevated in 9.8% (138/1,404) of men and 8.1% (69/852) of women. Of the 22 patients with abnormally low α-galactosidase A levels, 6 had abnormally high Gb3, but none had GLA gene variants. We found that none of the 19 patients with variants had abnormally low α-galactosidase A, while 9 of them had abnormal urine Gb3 levels (Figure 2). Interestingly, several of the missense mutations and complex intronic haplotypes that were identified have been reported in the medical literature to cause Fabry disease or Fabry-related symptomatology in specific cases.22,23,24,25 However, the male patients with these variants identified in this population did not have sufficiently low α-galactosidase A enzyme activity to result in clinical Fabry disease. About half of the patients identified were female, and α-galactosidase A activity was normal in this group, as is common even in females with Fabry disease due to the presence of the wild-type GLA allele. The frequencies of the exonic and intronic variants we identified in our patient population were no higher than their respective frequencies in the general population based on the 1000 Genomes (http://www.internationalgenome.org/data) or ExAc (http://exac.broadinstitute.org) databases (data not shown). The incidence of specific combinations of haplotypes in the general population is unknown.

Venn diagram of cardiac diagnoses in this study. CAD, coronary artery disease; Valve, valvular disease.

Venn diagram of cardiac patients with GLA gene variant abnormality ( GLA variant), abnormal α-galactosidase A activity (alpha GAL abnormally low), and elevated urine Gb 3 (Gb 3 abnormally high).
Discussion
Using three different screening methods, we found GLA sequence variants that have been previously reported to cause Fabry disease in certain patients. However, in this group of 2,256 patients with common heart disease, no previously undiagnosed patients with definitive Fabry disease were identified. Therefore, the cardiovascular common heart disease population, estimated to be approximately 85 million in the United States,26 is not likely to contain a sizable percentage of undiagnosed patients with Fabry disease; however, we could be expected to find some patients with undiagnosed Fabry disease in this population. It is likely that screening much larger cohorts or those selected high-risk populations with specific abnormalities such as idiopathic cardiomyopathy or patients on dialysis will provide a better diagnostic yield.17,27,28 Also, measurement of additional biomarkers, such as lyso-Gb3 and related analogues, may improve the sensitivity of methods for identifying Fabry disease.29
In this study, we used a general screening approach that is novel and may serve as a template for future screening studies of at-risk populations (e.g., idiopathic stroke, hypertrophic cardiomyopathy). We looked for Fabry disease at the level of the gene, the enzyme activity in blood, and the substrate in the urine in each patient, thus providing a unique opportunity to compare the different methods. We found that screening for Fabry disease using the sequencing of the whole GLA gene is the most useful method. This approach identifies pathogenic and benign variants of the GLA gene and provides a discriminating method to identify patients truly affected by Fabry disease.30,31 On the other hand, our study demonstrates that analysis of enzyme activity or the amount of substrate in the urine alone lacks the required specificity for large-scale screening. Urinary Gb3 was elevated in about 15% of patients with heart disease who otherwise did not have GLA mutations,20 and it is known to be normal in patients with mild mutations and in females,29 or is falsely elevated for other reasons.32 α-Galactosidase A activity was not a useful measurement either, because females often have normal enzyme levels33 and a substantial number (15) of males screened had enzyme activity near or below 2 μmol/L/hr (data not shown), a threshold found to contain patients with Fabry disease.34
The pathogenicity of the exonic variants that we found, R118C and D313Y, has been controversial. The R118C and D313Y variants were described in clinically diagnosed patients,35 in newborn screenings,2 in stroke in young patients, and in patients on renal dialysis in the Portuguese and Spanish populations, as well as in Brazilian patients. However, more comprehensive and critical investigations showed that these variants do not definitively lead to Fabry disease–related complications.31,36 D83N is a novel variant, but the patient had normal urine Gb3 levels and no other characteristics of Fabry disease. This variant was found to have high residual α-galactosidase A activity in the HEK-293 in vitro assay.37 However, assaying the enzyme activity in peripheral white blood cells of males is required to confirm non-pathogenicity. The intronic variants we found were described in some patients and may lower enzyme activity to a certain extent, but not enough to typically cause Fabry disease on their own, and elevated Gb3 was not present in skin biopsies (data not shown).38 A female with a Fabry-compatible phenotype who was heterozygous for a complex intronic haplotype has been reported, but the causality of this GLA gene abnormality was not conclusive.23
The combination of gene sequencing, assaying protein function, and substrate levels is an approach that may be applied to screening for other genetic/metabolic disorders. Such a strategy will allow not only determination of the most reliable initial screening method, but will serve to evaluate the pathogenicity of a particular novel genetic variation by assessing whether the genetic variant is associated with a significant reduction of protein function and an elevation of the related harmful metabolite. In our opinion, an enzymopathy like Fabry disease can be considered in a patient only when an abnormality is present in the entire biologic pathway, at least at the organ/tissue level.30
References
Meikle PJ, Hopwood JJ, Clague AE et al. Prevalence of lysosomal storage disorders. JAMA 1999;281:249–254.
Spada M, Pagliardini S, Yasuda M et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet 2006;79:31–40.
Mechtler TP, Stary S, Metz TF et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet 2012;379:335–341.
Hopkins PV, Campbell C, Klug T et al. Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J Pediatr 2015;166:172–177.
Linhart A, Lubanda JC, Palecek T et al. Cardiac manifestations in Fabry disease. J Inherit Metab Dis 2001;24:75–83; discussion 65.
Senechal M, Germain DP. Fabry disease: a functional and anatomical study of cardiac manifestations in 20 hemizygous male patients. Clin Genet 2003;63:46–52.
Sims K, Politei J, Banikazemi M et al. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke 2009;40:788–794.
Schiffmann R, Hughes DA, Linthorst GE et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 2017;91:284–293.
Scott LJ, Griffin JW, Luciano C et al. Quantitative analysis of epidermal innervation in Fabry disease. Neurology 1999;52:1249–1254.
Schiffmann R. Fabry disease. Pharmacol Ther 2009;122:65–77.
Brady R, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry disease: ceramide trihexosidase deficiency. N Engl J Med 1967;276:1163–1167.
Auray-Blais C, Cyr D, Ntwari A et al. Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease. Mol Genet Metab 2008;93:331–340.
Chatterjee S, Gupta P, Pyeritz RE et al. Immunohistochemical localization of glycosphingolipid in urinary renal tubular cells in Fabry’s disease. Am J Clin Pathol 1984;82:24–28.
Clarke JT, Guttmann RD, Wolfe LS et al. Enzyme replacement therapy by renal allotransplantation in Fabry’s disease. N Engl J Med 1972;287:1215–1218.
Winchester B, Young E. Biochemical and genetic diagnosis of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G (eds). Fabry Disease: Perspectives from 5 Years of FOS. Oxford PharmaGenesis: Oxford, UK, 2006, pp 169–181.
Schiffmann R, Waldek S, Benigni A et al. Biomarkers of Fabry disease nephropathy. Clin J Am Soc Nephrol 2010;5:360–364.
Sachdev B, Takenaka T, Teraguchi H et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002;105:1407–1411.
Kubo T, Ochi Y, Baba Y et al. Prevalence and clinical features of Fabry disease in Japanese male patients with diagnosis of hypertrophic cardiomyopathy. J Cardiol 2017;69:302–307.
Monserrat L, Gimeno-Blanes JR, Marin F et al. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2007;50:2399–2403.
Schiffmann R, Forni S, Swift C et al. Risk of death in heart disease is associated with elevated urinary globotriaosylceramide. J Am Heart Assoc 2014;3:e000394.
Harismendy O, Schwab RB, Bao L et al. Detection of low prevalence somatic mutations in solid tumors with ultra-deep targeted sequencing. Genome Biol 2011;12:R124.
Caetano F, Botelho A, Mota P et al. Fabry disease presenting as apical left ventricular hypertrophy in a patient carrying the missense mutation R118C. Rev Port Cardiol 2014;33:183 e181–185.
Pisani A, Imbriaco M, Zizzo C et al. A classical phenotype of Anderson-Fabry disease in a female patient with intronic mutations of the GLA gene: a case report. BMC Cardiovasc Disord 2012;12:39.
Zeevi DA, Hakam-Spector E, Herskovitz Y et al. An intronic haplotype in alpha galactosidase A is associated with reduced mRNA expression in males with cryptogenic stroke. Gene 2014;549:275–279.
Tuttolomondo A, Duro G, Pecoraro R et al. A family with various symptomatology suggestive of Anderson-Fabry disease and a genetic polymorphism of alpha galactosidase A gene. Clin Biochem 2015;48:55–62.
Mozaffarian D, Benjamin EJ, Go AS et al. Executive summary: heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation 2016;133:447–454.
Linthorst GE, Bouwman MG, Wijburg FA et al. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet 2010;47:217–222.
Saito O, Kusano E, Akimoto T et al. Prevalence of Fabry disease in dialysis patients: Japan Fabry disease screening study (J-FAST). Clin Exp Nephrol 2016;20:284–293.
Auray-Blais C, Lavoie P, Boutin M et al. Biomarkers associated with clinical manifestations in Fabry disease patients with a late-onset cardiac variant mutation. Clin Chim Acta 2017;466:185–193.
Schiffmann R, Fuller M, Clarke LA et al. Is it Fabry disease? Genet Med 2016;18:1181–1185.
Ferreira S, Ortiz A, Germain DP et al. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: data from individual patients and family studies. Mol Genet Metab 2015;114:248–258.
Gaggl M, Lajic N, Heinze G et al. Screening for Fabry disease by urinary globotriaosylceramide isoforms measurement in patients with left ventricular hypertrophy. Int J Med Sci 2016;13:340–346.
Linthorst GE, Vedder AC, Aerts JM et al. Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. Clin Chim Acta 2005;353:201–203.
Scott CR, Elliott S, Buroker N et al. Identification of infants at risk for developing Fabry, Pompe, or mucopolysaccharidosis-I from newborn blood spots by tandem mass spectrometry. J Pediatr 2013;163:498–503.
Eng CM, Resnick-Silverman LA, Niehaus DJ et al. Nature and frequency of mutations in the alpha-galactosidase A gene that cause Fabry disease. Am J Hum Genet 1993;53:1186–1197.
Arends M, Wanner C, Hughes D et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol 2017;28:1631–1641.
Benjamin ER, Della Valle MC, Wu X et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med 2017;19:430–438.
Gervas-Arruga J, Cebolla JJ, Irun P et al. Increased glycolipid storage produced by the inheritance of a complex intronic haplotype in the alpha-galactosidase A (GLA) gene. BMC Genet 2015;16:109.
Acknowledgments
This work is supported in part by Baylor Research Institute Foundation, Amicus Therapeutics, Inc., and the National Institutes of Health U54 NS065768 (project LDN 6711). The Lysosomal Disease Network (U54NS065768) is part of the Rare Diseases Clinical Research Network, an initiative of the Office of Rare Diseases Research, and the National Center for Advancing Translational Sciences. This consortium is funded through collaboration between the National Center for Advancing Translational Sciences, the National Institute of Neurological Disorders and Stroke, and the National Institute of Diabetes and Digestive and Kidney Diseases. We thank David J. Lockhart for his support of this study and key insights and Marie-Anne Schiffmann for editing and proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
R.S. has received research funding from Amicus Therapeutics, Protalix Biotherapeutics, Shire, and Sanofi Genzyme. E.R.B., J.P.C., J.B., and X. Wu are employees of Amicus Therapeutics. The other authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Schiffmann, R., Swift, C., McNeill, N. et al. Low frequency of Fabry disease in patients with common heart disease. Genet Med 20, 754–759 (2018). https://doi.org/10.1038/gim.2017.175
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.175
This article is cited by
-
Phenotypic Evolution in Fabry Disease: Our Experience in Indian Cohort
Indian Journal of Clinical Biochemistry (2024)
-
The prevalence of Fabry disease among 1009 unrelated patients with hypertrophic cardiomyopathy: a Russian nationwide screening program using NGS technology
Orphanet Journal of Rare Diseases (2022)
