
Abstract
Purpose: Bardet-Biedl syndrome is a pleiotropic multiple anomaly syndrome inherited in an autosomal recessive pattern. It is now known that this disorder has locus heterogeneity, with causative mutations identified in as many as 14 genes. The aim of this study was to derive locus-specific recurrence risk estimates for family members of a proband affected with Bardet-Biedl syndrome.
Methods: Mutation data from 187 probands affected with Bardet-Biedl syndrome were used. The authors counted the relative proportion of families with mutations at each of 10 loci and estimated locus-specific carrier rates for mutations using Hardy-Weinberg principles and an aggregate population frequency of 1/100,000 for the phenotype. Locus-specific recurrence risks were calculated for relatives of an affected proband.
Results: Locus-specific carrier frequencies range from 1/250 to 1/2200, and the risks for an offspring of the sibling of an affected individual range from 1/1,500 to 1/13,000. The estimate of this risk derived under a locus homogeneity model is 1/960.
Conclusion: Variation of recurrence risks of this magnitude may have implications for genetic counseling of families with affected individuals, in particular about prenatal testing and other reproductive options. Similar analyses to determine locus-specific carrier frequencies for other phenotypes with significant locus heterogeneity may yield similarly relevant results.
Similar content being viewed by others

Main
The clinical entity of Bardet-Biedl syndrome (BBS) was described nearly 90 years ago1 and comprises primarily of polydactyly, obesity, pigmentary retinopathy, cognitive impairment, and renal and genitourinary anomalies. In the past 9 years, remarkable progress has been made in the elucidation of the genetic etiology of this disorder2–13 and the underlying pathophysiology of the primary cilium.14,15 These molecular discoveries are improving clinical care and may eventually provide specific therapeutic opportunities for patients with BBS.
Clinical practitioners are increasingly called on to evaluate, molecularly diagnose, and counsel individuals and families affected by phenotypes suggestive of BBS. In our experience, parents of children affected with BBS are quite concerned about the risks for their unaffected offspring to have children with the disorder, and they report that more distant relatives share this concern (unpublished data). The discovery and elucidation of the remarkable locus and allelic heterogeneity of BBS has implications for clinical practice because it affects the calculation of risks for family members (other than the recurrence risk for parents of a child affected with BBS, which in nearly all cases is 1/4). Furthermore, the carrier frequency for a given locus can be significantly lower than that predicted by the frequency of the phenotype. Here, we report a survey of mutations among BBS loci and derive recurrence risk figures for relatives other than full siblings for 10 BBS loci.
MATERIALS AND METHODS
We outline here our approach to estimating gene-specific carrier frequencies along with the data and calculations we used to arrive at our estimates of recurrence risks for relatives of an individual with BBS to have a child with BBS. We present this approach using as examples the risks that would pertain to a hypothetical first offspring of various relatives of a patient with BBS, assuming that those individuals reproduced with an unrelated spouse. A number of assumptions were made for this analysis. First, we assumed that BBS is inherited in an autosomal recessive pattern (in agreement with Ross and Beales16). This assumption implied that occurrences of oligogenic17 and apparently digenic BBS18 are sufficiently rare that they can be set aside for this analysis. Second, we estimated an overall population occurrence rate of 1/100,000 for the BBS phenotype.14 We assumed that this number is relevant to a general North American outbred population, descended primarily from Europeans. It specifically excluded subgroups such as the Old Order Amish (shown to have a high frequency of McKusick Kaufman syndrome caused by mutations in MKKS19), the Newfoundland population (which also has a high prevalence of MKKS mutations that cause BBS11,20), other groups who are of geographically distinct origins, and groups or families with known consanguinity. Third, we estimated the relative proportion of mutant BBS alleles among the BBS loci by counting probands who have been found to have two pathogenic alleles in a single BBS gene. We reasoned that identifying mutant alleles through affected probands was a conservative approach, because the prior probability was very small that a patient with BBS would have two rare variants in a single gene without that gene being causative of the phenotype in that patient. Although we cannot directly determine the actual prevalence of mutant alleles from such an approach, we reasoned that this approach would allow us to determine the relative contributions of BBS loci to the overall frequency of mutant BBS alleles, which would then allow us to estimate the relative contribution of each locus to the overall prevalence of the disorder.
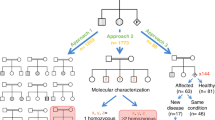
To make an estimate of the distribution of alleles among the BBS loci, we used data from three cohorts of patients: The Sick Children's Hospital cohort in Toronto, the University of Iowa Hospitals and Clinics cohort, and the National Human Genome Research Institute (NHGRI) cohort in Maryland. We excluded from this analysis cases in which it was known that the family was of non-European geographic origin. The mutations were determined by standard methods in each of three research laboratories. Briefly, DNA was isolated from peripheral blood leukocytes, polymerase chain reaction-amplified, and subjected to dideoxy terminator sequencing. Although the approach varied somewhat in each of the laboratories, the methods were similar in that each DNA sample was sequenced for the more common genes first (e.g., BBS1 and BBS108,12) followed by additional sequencing of other BBS genes until two mutations in a single gene were identified. For the purpose of this study, sequence variants were designated as mutations if two variants were found in one BBS gene, they were not recognized as a common variant, and other analyses (e.g., evolutionary conservation, proper phase, etc.) supported the notion that they were deleterious. Missense alterations were considered deleterious based on amino acid conservation and absence in controls, and each mutation was confirmed on a second polymerase chain reaction product. Each proband was counted as a case; other affected members of that family were not counted. MKS1 and CEP290 were not analyzed in these cohorts because causation has yet to be confirmed for these loci. We excluded TTC8 and TRIM32 as no cases with two mutations at these loci were identified in our cohorts. We estimated the carrier rate and the chance that a given individual in a hypothetical pedigree (Fig. 1) would be affected, first assuming locus homogeneity and then under a model of locus heterogeneity with unequal locus frequencies based on our mutation data (reviewed by Sheffield and coworkers).14

Hypothetical pedigree of a family of a woman affected with BBS. Locus-specific risk estimates derived from the carrier frequencies calculated in Table 1 for each first offspring of multiple possible matings are shown along with estimates based on a locus homogeneity (LH) model.
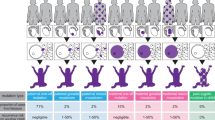
We used Hardy-Weinberg principles, starting from the overall population prevalence of BBS and using the relative proportions of affected probands at each of 10 loci to estimate the mutant allele frequency for each locus. We then used the carrier rates to calculate familial recurrence risks, using the mutant allele frequency to estimate the likelihood that the married in, unrelated parent was a carrier. Decimal values of carrier frequency were transformed into fractions with two significant digits in the denominator and these rounded fractions were used to calculate specific recurrence risk estimates. The mutation research studies were reviewed and approved by the research ethics committees at the three participating institutions.
RESULTS
We calculated locus-specific risk estimates for each gene for a relative of a patient with BBS and compared these estimates to that derived from the locus homogeneity model. Table 1 shows our carrier frequency calculations and the locus-specific risk estimate calculations for individual IV-2 in Figure 1 (the niece/nephew of an affected individual). These estimates are expanded for other relatives in Figure 1.
Under a locus homogeneity model, we reasoned that q2 = 1/100,000, and therefore q ≅ 0.0032; the carrier frequency would then be estimated to be 2 pq ≅ 0.0032 × 2 = 0.0063 (0.63% or 1/160). Thus, the risk for a full sibling of an affected individual to have an affected child would be estimated to be 0.67 × 0.0063 × 0.25 = 0.0011, or approximately 1/960.
A total of 187 index cases were ascertained among the three laboratories (Table 1). The data among the three laboratories support prior data in that BBS1 and BBS10 were more commonly mutated than all other loci, comprising almost 70% of the families. However, the Iowa cohort included more families with BBS1 mutations than for BBS10. The risk for hypothetical individual IV-2 associated with the other 10 loci ranged from ∼1/1,500 for BBS1 to ∼1/13,000 for ARL6.
DISCUSSION
Our experiences in caring for families affected with BBS and other disorders inherited in an autosomal recessive pattern suggest to us that a change in recurrence risk of less than 2-fold is unlikely to be clinically significant, especially when the risks discussed fall well below 1/100. In this context, our experience suggests that modifying, for example, the recurrence risk estimates for the offspring of a sibling of an affected individual in a family with ARL6 mutations from 1/960 to 1/13,000 is likely to be clinically relevant. A risk modification of this magnitude may be especially salient when relatives of affected individuals present for genetic counseling and the consideration of carrier testing is discussed. In contrast, these estimates may have little impact for a family with an affected member with mutations in BBS1 (a risk change from 1/960 [locus homogeneity] to 1/1500 [locus-specific]). Similarly, family members of individuals with BBS10 mutations may or may not find the approximate halving of their risk to be significant. In such cases, other factors, such as the family's lived experience with BBS, number of affected children within a sibship, general views of disability, etc., are likely to have a greater impact than this degree of change in numerical risk.21 Regardless of the magnitude of the risk, it should be presented in the context of the at-risk couple's background risk for all serious birth defects, which ranges from 2 to 3%.
These risk calculations may also be relevant for patients affected with BBS. Although many affected individuals may encounter social or medical (e.g., hypogenitalism and spermatozoa abnormalities in males and genitourinary malformations in females) impediments to reproduction, affected individuals have had children and it is possible that they might have concerns about the risk of having a child affected with BBS. In fact, there has been one report in the literature of successful reproduction22 and one female patient in the NHGRI cohort has reproduced (unpublished data). Thus, we estimated the risks for offspring of an affected individual under both the locus homogeneity and locus-specific model in Figure 1.
These estimates are subject to several limitations in the data presented and the analysis performed here. First, the methods used to estimate relative prevalence are limited by the number of cases evaluated at the three centers, referral and ascertainment biases, and the possible existence of families with mutations that are difficult to detect by standard methods (which may be unevenly distributed across the loci). The limitation of the number of cases is especially relevant with respect to the rare loci. Although we are confident that BBS1 and BBS10 mutations are much more commonly found in patients with BBS than are mutations in the other loci, the magnitude of this difference is imprecise. For example, only one case of a family with an ARL6 mutation was identified. The addition of one additional family with mutations at this locus would significantly change the risks for that locus. Thus, more cases are needed and periodic reassessment of the relative contribution of each locus to the proportion of mutant alleles is warranted. Second, our method of case finding (using cases with two recognizable mutations) would miss families with atypical mutations, which again may be unevenly distributed across the loci, distorting the recurrence risks. These risk calculations are less relevant for persons who are of non-European origin for two reasons. Our sample sets did not include a sufficient number of cases of persons in those groups and for some groups (e.g., the Bedouins), their recurrence risks are influenced little by locus heterogeneity but more by the degree of consanguinity of the spouses.
The data reported here do not have utility in the risk assessment for a couple within a BBS family regarding their risk for having an affected child before molecular testing is performed; and the risk calculations presented here require a molecular diagnosis. For patients who require a recurrence risk before molecular testing, we recommend using risks based on the common loci. As BBS1 and BBS10 are the most frequent loci and their carrier frequencies do not differ dramatically (1/250 for BBS1 and 1/290 for BBS10), we suggest that in situations where risks need to be estimated before testing, basing recurrence risk estimates on the carrier frequency associated with BBS1 mutations is likely to be most clinically relevant and approach the highest degree of accuracy for many families, while it over-estimates risk for a minority of families. This is preferable to using the locus homogeneity risk because this will falsely elevate the risk for relatives other than the affected individual's full siblings.
Our analysis does not have utility for couples from families where the individual affected with BBS has tested negative for BBS mutations at the known loci. To our knowledge, molecular testing for BBS mutations in the United States is commercially available for only three loci (BBS1, BBS2, and BBS10). Recurrence risk estimates based on the locus homogeneity model for families who undergo commercially available testing that fails to find mutations would be likely to over estimate their risk. We suggest that in these cases, referral to a research group for clinically validated sequencing of the remaining genes may be warranted. The advent of affordable whole genome and exome sequencing will dramatically change this situation. Such technologies will allow for rapid and complete interrogation of all genes and will require more sophisticated risk estimations that incorporate factors such as compound heterozygosity and potential modifiers.
Despite the limitations of our analysis, it is clear that the locus-specific recurrence risk for having an affected child for relatives of individuals affected with BBS (other than those within the full sibship of the affected individual) is lower than that which would be estimated from the aggregate prevalence or incidence rates of the phenotype. For the two common loci, the magnitude of that difference is modest, and for the rare ones, it is large and likely clinically relevant. Similar analyses to determine locus-specific carrier frequencies for other phenotypes with significant locus heterogeneity (e.g., nonsyndromic autosomal recessive hearing loss23) may yield similarly relevant results. Finally, we note in Table 1 that these estimates predict a 2.67-fold higher aggregate carrier frequency for the multilocus model (1.7%) when compared with the estimate based on locus homogeneity (0.63%). This high aggregate carrier frequency suggests to us the coexistence of a heterozygous, or third mutation in a second BBS gene is not a rare event in patients with BBS.
References
Bardet G . Sur un syndrome d'obésité congenitale avec polydactylie et rétinité pigmentaire (contribution a l'étude des formes cliniques de l'obésité hypophysaire). Paris, Universite de Paris, 1920.
Ansley SJ, Badano JL, Blacque OE, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 2003; 425: 628–633.
Badano JL, Ansley SJ, Leitch CC, Lewis RA, Lupski JR, Katsanis N . Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am J Hum Genet 2003; 72: 650–658.
Chiang AP, Beck JS, Yen HJ, et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc Natl Acad Sci U S A 2006; 103: 6287–6292.
Chiang AP, Nishimura D, Searby C, et al. Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3). Am J Hum Genet 2004; 75: 475–484.
Li JB, Gerdes JM, Haycraft CJ, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 2004; 117: 541–552.
Mykytyn K, Braun T, Carmi R, et al. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet 2001; 28: 188–191.
Mykytyn K, Nishimura DY, Searby CC, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet 2002; 4: 429–437.
Nishimura DY, Searby CC, Carmi R, et al. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2). Hum Mol Genet 2001; 10: 865–874.
Nishimura DY, Swiderski RE, Searby CC, et al. Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am J Hum Genet 2005; 77: 1021–1033.
Slavotinek AM, Stone EM, Mykytyn K, et al. Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet 2000; 26: 15–16.
Stoetzel C, Laurier V, Davis EE, et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat Genet 2006; 38: 521–524.
Stoetzel C, Muller J, Laurier V, et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet 2007; 80: 1–11.
Loktev AV, Zhang Q, Beck JS, et al. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell 2008; 15: 854–865.
Badano JL, Mitsuma N, Beales PL, Katsanis N . The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 2006; 7: 125–148.
GeneReviews. Review. 2009. Available at: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=bbs#bbs.grID34742. Accessed December 30, 2009.
Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 2001; 293: 2256–2259.
Fauser S, Munz M, Besch D . Further support for digenic inheritance in Bardet-Biedl syndrome. J Med Genet 2003; 40: e104.
Stone DL, Slavotinek AS, Bouffard GG, et al. Mutations of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome. Nat Genet 2000; 25: 79–82.
Katsanis N, Beales PL, Woods MO, et al. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat Genet 2000; 26: 67–70.
Shiloh S, Saxe L . Perception of risk in genetic counseling. Psychol Health 1989; 3: 45–61.
Iannello S, Bosco P, Cavaleri A, Camuto M, Milazzo P, Belfiore F . A review of the literature of Bardet-Biedl disease and report of three cases associated with metabolic syndrome and diagnosed after the age of fifty. Obes Rev 2002; 3: 123–135.
Petersen MB, Willems PJ . Non-syndromic, autosomal-recessive deafness. Clin Genet 2006; 69: 371–392.
Acknowledgements
Supported by research funding from the intramural program of the National Human Genome Research Institute (to JCS, JJJ, and LGB). Supported by NIH (R01-EY-11298) (to VCS and EMS). Supported by the Foundation Fighting Blindness-Canada (to EH).
The authors thank Katie Wetsch and Emma Spaulding for assistance with the sequencing of BBS genes, Julia Fekecs and Danielle Brinckman for graphics assistance, and Barbara Biesecker for critical review of the manuscript. The authors dedicate this paper to Dr. Penelope Philbrick Feuillan, a talented clinician and fellow BBS researcher, who died November 7, 2008.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: VCS and EMS are investigators of the Howard Hughes Medical Institute. The remaining authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Sapp, J., Nishimura, D., Johnston, J. et al. Recurrence risks for Bardet-Biedl syndrome: Implications of locus heterogeneity. Genet Med 12, 623–627 (2010). https://doi.org/10.1097/GIM.0b013e3181f07572
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181f07572
