
Abstract
Purpose: Based on results of diagnostic MEN1 testing, we have attempted to further define the mutational spectrum of the MEN1 gene and the clinical features most frequently associated with MEN1 mutations.
Methods: Mutation testing was performed on blood samples by PCR amplification and sequencing of exons 2 to 10 of the MEN1 gene and the corresponding intron-exon junctions. Pedigree phenotypic information was obtained by written questionnaire.
Results: Among 288 presumably unrelated pedigrees, 73 independent mutations were found in 89 families. Five mutations were found in 2 pedigrees, and 4 mutations were seen in more than 2 pedigrees. There were 17 nonsense mutations (23.3%), 2 in-frame deletions (2.7%), 18 frameshift-deletion mutations (24.7%), 10 frameshift-insertion or -duplication mutations (13.7%), 13 splice-site mutations (17.8%), and 13 presumptive missense mutations (17.8%). Thirty-nine of 56 pedigrees with parathyroid and pancreatic islet neoplasia tested positive, compared with 4/24 and 8/32 pedigrees affected with hyperparathyroidism or hyperparathyroidism and pituitary tumors. MEN1 mutations were found in 6/20 sporadic patients, all of whom had both parathyroid and pancreatic neoplasms. Of 14 mutation-negative sporadic patients, 10 exhibited hyperparathyroidism and pituitary tumors without islet cell neoplasia. Somatic mosaicism was detected in 1 sporadic patient.
Conclusion: Patients from pedigrees with hyperparathyroidism and pancreatic islet tumors are most likely to test positive for MEN1 mutations. Mutations are less often detected in patients from pedigrees with hyperparathyroidism alone or in combination with pituitary tumors without pancreatic islet neoplasia. Sporadic cases are less likely to test positive than familial cases, in part due to somatic mosaicism.
Similar content being viewed by others

Main
MEN1 is an autosomal dominant disorder that is characterized by parathyroid (95%–100%), benign and malignant pancreatic islet (35%–75%), and anterior pituitary neoplasms (15%–40%). Foregut carcinoid tumors (thymic and bronchial), adrenocortical hyperplasia, multiple lipomas, facial angiofibromas, and skin collagenomas may also be seen in the syndrome.1–4 Unusual features of MEN1-related tumors include their multiplicity, both within and among target organs, and an earlier age of onset than similar sporadic tumors.5
Symptoms in MEN1 are caused by an overproduction of specific hormones, neoplasm mass effects, or malignant transformation. Although most tumors in MEN1 patients are benign, pancreatic islet and carcinoid neoplasms have significant malignant potential and are currently the most important cause of MEN1-related mortality.3,6 Based on biochemical data, the prevalence of MEN1 is estimated at 10 to 175 per 1,000,000 individuals, although autopsy findings suggest that the frequency of occurrence may be as high as 25 per 10,000.7 The clinical expression of MEN1 generally manifests in the third to fourth decade, with nearly complete penetrance by the early fifties. Clinical disease is uncommon before 10 years of age.1,3,8
The MEN1 gene, located at chromosome 11q13, is approximately 9.8 kb in length and contains 10 exons. The 1830-bp coding region in exons 2 to 10 generates a 610 amino acid protein of unknown function called “menin.” Menin appears to reside primarily in the nucleus and bears no homology to other known proteins. It is expressed in a wide range of tissues and is conserved through evolution from Drosophila to humans. Because MEN1-related tumors demonstrate loss of heterozygosity (LOH) of the chromosome 11q13 region, menin has been proposed to function as a tumor suppressor in accordance with the Knudson “two-hit” hypothesis.9–13 However, several investigations suggest that MEN1 may be involved in the maintenance of genomic integrity in a manner similar to that of BRCA1 and BRCA2 or the genes underlying HNPCC, which also show LOH in tumors.14,15
The discovery of the MEN1 gene by positional cloning in 1997 resulted in the development of genetic tests for responsible mutations.16,17 Dispersed nonsense, missense, frameshift, in-frame deletions, and splice-site mutations have been described in the syndrome, with the majority of pedigrees having unique mutations. More than 300 mutations in the MEN1 gene have been reported, but no definite genotype-phenotype correlations have been shown.7,18–21
DNA-based testing for MEN1 provides a means for more accurate diagnosis. Medical monitoring for signs and symptoms of MEN1-related neoplasms in at-risk individuals can lead to early interventions that are predicted to positively influence the course of the disease. Genetic testing has been recommended for index cases who meet clinical criteria for familial or sporadic MEN1, for those who do not meet formal MEN1 criteria but are suspicious for the syndrome, and for symptomatic or asymptomatic relatives of patients with known MEN1 mutations. Positive test results can alert presymptomatic MEN1 mutation-positive individuals to the need for monitoring, whereas negative tests allow members of affected families to avoid costly, time-consuming, and potentially psychologically distressing screening. Genetic testing can also assist members of MEN1-affected kindreds in family planning.22
In this study, we report the results of clinical MEN1 testing in a DNA diagnostic laboratory, including data that can assist patient counseling and help direct appropriate test ordering. Because samples and clinical data from patients were not obtained through a systematic scheme, inaccuracies may have been introduced into some of our results. This objection notwithstanding, the data are valuable because they reflect the nature, extent, and quality of information likely to be encountered by “real-world” practitioners. Furthermore, the utility of this study is suggested by the concordance of our results and conclusions with those of other investigations with more systematic ascertainment and clinical evaluation of study subjects.
MATERIALS AND METHODS
Participants
From October 1997 to May 2003, blood samples from 400 individuals representing 288 pedigrees located primarily in the United States and Canada were received in the Yale DNA Diagnostic Laboratory for MEN1 mutation testing. A request for clinical information, including patient and family history, was included as a part of our laboratory's sample requisition form. In addition, a one-page patient and family history questionnaire was mailed to the referring physician or genetic counselor for each patient about whom sufficient clinical information did not accompany the sample. Clinical information was obtained for at least one member of 144 pedigrees and a total of 200 patients.
Mutation testing
Genomic DNA from peripheral blood leukocytes was extracted using standard methods.23,24 DNA sequence abnormalities were identified by PCR amplification and sequencing of the 9 coding exons of the MEN1 gene and the corresponding intron-exon junctions as previously described.25 All mutations were confirmed by bidirectional sequencing, or by the use of restriction enzyme digests for mutations that were predicted to alter restriction sites. Potential splice-site mutations that altered conserved but not invariant bases were confirmed by RT-PCR analysis of MEN1 mRNA. In-frame deletions and missense mutations were judged deleterious if they were not found in 200 unaffected individuals, altered highly conserved base pairs, and segregated with the disease in those cases for which more than one affected family member was available for study. Once the disease causing mutation was identified in a family, additional family members were tested by direct sequencing or restriction enzyme digestion.
In one sporadic patient in whom sequencing suggested mosaicism for a nonsense mutation, PCR products from the affected exon were TA cloned (Invitrogen, Carlsbad, CA) and directly sequenced.
Determining the origin of a recurrent mutation
Haplotyping around a recurrent 249delGTCT mutation was performed by PCR amplification of a microsatellite locus at the human muscle glycogen phosphorylase gene (PYGM) a polymorphic marker that lies approximately 55 kb 3′ to the MEN1 gene,16,20,26 and a CA repeat in the region of the MEN1 gene 1680 bp 5′ to the start of exon 1 that was developed as part of this study. The primer sequences used to amplify this latter marker were as follows: F cgcctaattttgtgtgtatg; R agctgggaatccctgtctctg.
Statistical analysis
The statistical significance of differences in the proportions of patients testing positive for MEN1 mutations among various phenotypes was assessed using the chi-square test or Fisher exact test.
RESULTS
Eighty-nine out of the 288 presumably unrelated pedigrees demonstrated MEN1 mutations. Among the 89 MEN1-positive pedigrees, 73 independent mutations were identified (Table 1). These mutations were scattered throughout the coding region, with the proportion of mutations found in each exon roughly correlating with exon size (Fig. 1). Most mutations detected produce truncated proteins. Seventeen nonsense mutations (23.3%), 2 in-frame deletions (2.7%), 18 frameshift-deletion mutations (24.7%), 10 frameshift-insertion or -duplication mutations (13.7%), 13 splice-site mutations (17.8%), and 13 presumptive missense mutations (17.8%) were identified.

Mutation distribution by exon. Mutations were found within exons at frequencies that were roughly proportional to exon size.
Five mutations were found in 2 pedigrees, and 4 were seen in more than 2 pedigrees (Fig. 2). The most common mutation identified was a previously described 4-bp deletion in codons 83 to 84 (249delGTCT) that was present in 6 (6.7%) of the affected pedigrees.8 Haplotyping around the 249delGTCT mutation in affected representatives of these families demonstrated a lack of relatedness, predicting multiple independent origins for the mutation (Table 2).

Recurrent mutations. Majority of the 89 MEN1-positive pedigrees had independent mutations. Five mutations were found in two pedigrees, and 4 mutations were seen in more than 2 pedigrees (see also Table 2). The most common mutation identified was a previously reported 4-bp deletion in codons 83 to 84, 249delGTCT, that was present in 6 (6.7%) of the affected pedigrees.
Among the 144 kindreds for whom clinical data were available, those most likely to test positive had parathyroid and pancreatic islet neoplasia, whether or not family members also had pituitary or other tumors (39/56 pedigrees) (Table 3). There was no significant difference between the proportion of positive tests in families with parathyroid, pancreatic islet, and pituitary tumors (22/30) and families with parathyroid and pancreatic islet tumors without pituitary tumors (17/26; P = 0.52). Pedigrees with the combination of hyperparathyroidism and pituitary tumors but without pancreatic islet neoplasia were less likely to test positive for MEN1 mutations (8/32) than families with any combination of the key MEN1 neoplasms including parathyroid and pancreatic islet tumors (P < 0.0001). Likewise, families were less likely to test positive if they had isolated hyperparathyroidism (4/24, P < 0.001), pancreatic islet (2/10, P = 0.005), or pituitary tumors (3/10, P = 0.03). The combination of pituitary and pancreatic islet tumors without hyperparathyroidism was too infrequent for comparisons to yield statistically significant results.
Among the 144 kindreds, there were 20 that involved only a single affected patient with no other affected family members. These sporadic patients had tumors in at least two of the three major tissues affected in MEN1 (Table 4). Among patients who had hyperparathyroidism and pancreatic islet tumors, with or without other tumors, 6/10 tested positive. Among the remaining 10 sporadic patients, all of whom had hyperparathyroidism in combination with pituitary tumors, none tested positive (P = 0.01).
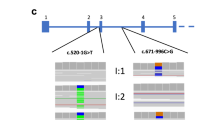
One of the sporadic patients, a 27-year-old woman with hyperparathyroidism, a prolactinoma, and a pancreatic mass, had a possible CAG→TAG mutation in codon 405 of exon 9 (Q405X) with gene dosage for the abnormal base that was significantly < 50% (Fig. 3). Somatic mosaicism was investigated in the patient by cloning the exon 9 PCR products, and 2 out of 20 clones were found to contain the Q405X mutation.

Somatic mosaicism in a sporadic patient. A, Sequence from peripheral blood leukocyte DNA showing low level of mosaicism for the Q405X mutation in exon 9. First base in codon 405 is highlighted. Note reduced peak height for C compared with the normal sequence and an underlying T peak. B, Exon 9 PCR products were TA cloned and sequenced. Two of out 20 clones showed the Q405X mutation. C, Normal comparison sequence.
DISCUSSION
Clinical disease in MEN1 patients may be the result of hormone hypersecretion, tumor mass effects, malignancy, or any combination of these. Approximately one-third of deaths in MEN1 are caused by associated malignancies. Particularly because of the malignant potential of MEN1-related neuroendocrine tumors of the thorax, pancreas, and duodenum, and because of the early age of onset and possibly greater aggressiveness of pituitary tumors in the setting of MEN1, genetic testing and annual biochemical screening has been advocated in individuals known to be at risk for MEN1 mutations, beginning in childhood.22,27
Selected imaging studies should be performed every 3 to 5 years in those who exhibit biochemical evidence of neoplasia or display signs and symptoms of an MEN1-related tumor. Biochemical markers may include total or ionized calcium, intact parathyroid hormone, fasting glucose and insulin, prolactin, and IGF-1 in children, with testing for fasting gastrin, glucagon, proinsulin, pancreatic polypeptide, and chromogranin A levels among others incorporated at age 20. Biochemical screening can detect the presence of MEN1-related neoplasms at least 5 to 10 years before clinically evident disease is present.1,3,8,22,27,28
Our laboratory began offering clinical testing for germline mutations in the MEN1 gene shortly after its discovery in 1997. Genetic testing of asymptomatic, at-risk patients who do not possess the mutations found within their families allows MEN1-negative individuals to be spared regular biochemical evaluations and provides reassurance that they are not at risk to pass on the disease to their progeny. Conversely, the presence of an MEN1 mutation suggests the need for biochemical and radiologic monitoring, which is predicted to have beneficial effects on the disease course.2,3,8,22,28–32
Slightly > 30% of the 288 pedigrees that we tested were found to have MEN1 mutations. The mutation detection rate in clinically diagnosed familial MEN1 patients is believed to be in the range of 80% to 90% with a somewhat lower detection rate for sporadic cases. The comparatively low percentage of MEN1-positive families in our test population is to be expected because many pedigrees did not meet standard clinical criteria for familial MEN1: a proband with at least two neoplasms in major MEN1-related tissues (parathyroids, anterior pituitary, or pancreatic islets) and at least one additional first degree relative with an MEN1-related endocrinopathy.7 Among pedigrees strictly defined as having familial MEN1, we found mutations in 76.3%, which approaches the rates reported by others.8,20,22,26,33,34
In pedigrees that do meet the diagnostic criteria for familial MEN1, a failure to find mutations in the MEN1 gene could reflect the presence of functionally significant intronic mutations that would not be detected with currently used primer sets, mutations in regulatory or important nonintronic untranslated regions, disease phenocopies not caused by mutations in MEN1, or large deletions that would be missed by gene sequencing.35 Large deletions have been reported in only 4 families although over 300 mutations have been reported in the medical literature and the Human Gene Mutation Database.36 Nevertheless, such deletions may be more common in MEN1 than previously believed, and our yield may be increased by the addition of Southern blotting or other gene dosage techniques to our standard procedures. Finally, we provide evidence that somatic mosaicism can account for some sporadic cases of MEN1. MEN1 mutations in such patients are considerably less likely to be detected by sequencing than mutations in nonmosaics.
Consistent with previous reports, the mutational spectrum was broad among the MEN1 pedigrees that we tested,8,16,20,21,26,28,37 and the majority of mutations that we detected have not been previously described. Most were predicted to result in loss of menin function due to premature truncation of the gene product.
Several recurrent mutations were found in apparently unrelated pedigrees, suggesting founder effects or the presence of mutational hotspots in the gene. Haplotyping around a previously described 4-bp deletion in codons 83to 84 of the MEN1 gene indicated that this mutation arose independently multiple times. Together with published data, our analysis establishes codons 83 to 84 as a mutational hotspot. A replication-slippage model has been postulated for the increased mutation rate in these codons based upon the presence of CT dinucleotide repeats in the flanking regions of the deletion.8,37
Among the samples submitted to our laboratory, the best predictor of a positive test was a personal or family history of parathyroid and pancreatic islet neoplasia with or without other tumors. The combination of pancreatic islet neoplasia with pituitary tumors was probably also a good predictor of a positive test, but this category was too small to draw statistically significant conclusions. Families with isolated hyperparathyroidism, isolated pancreatic islet tumors, or isolated pituitary tumors were significantly less likely to test positive, but still in a range that would typically justify testing for a hereditary cancer syndrome. None of those with carcinoid alone, or adrenal cortical hyperplasia alone, tested positive; but there were too few cases to make definitive recommendations about MEN1 testing in these groups.
Although still meriting genetic testing, families with the combination of pituitary and parathyroid neoplasia were much less likely to test positive than those with a combination of neoplasms that included the pancreatic islets. None of 10 sporadic patients with parathyroid and pituitary neoplasia tested positive (95% confidence intervals on this proportion range from 0% to 30%). The relative infrequency of MEN1 mutations in sporadic patients who exhibit the combination of hyperparathyroidism and pituitary tumors is consistent with previous literature reviewed.38 Given the substantial incidences of parathyroid and pituitary tumors in the U.S. population, the presence of these neoplasms in a pedigree may be explained by random chance. However, the likelihood of an individual developing both pituitary and parathyroid tumors is considerably less. Thus, the relatively limited number of MEN1-positive pedigrees with parathyroid and pituitary tumors but without islet tumors, combined with the large proportion of sporadic cases with this constellation of tumors, raises the possibility that the phenotype frequently represents a different disease. The sporadic patients in this setting could be explained by autosomal recessive inheritance, or by a relatively high rate of de novo mutations in a putative autosomal dominant gene. Conceivably, specific mutations in MEN1 not detected by sequencing exons 2 to 10 could account for this phenotype.
This review represents an analysis of the characteristics and test results of all patients referred to our laboratory for MEN1 testing during the study period. Although the method of ascertainment should not specifically introduce bias into the results, one interesting observation is that those families about whom we obtained clinical data were more likely to test positive for MEN1 than pedigrees from which no clinical information available (59/144 vs. 30/144; P < 0.0002). A significant number of patient samples from kindreds in this latter category were referred as send-out tests from large reference laboratories or hospital laboratories. The significance of this observation is unclear.
Because our results are those of patient samples sent for clinical testing, as opposed to research subjects, we had little influence over the amount or quality of clinical information that we received about each pedigree. The lack of standardized clinical assessment among patients and pedigrees could have lead to some inaccuracy in the data. For example, a failure to meticulously evaluate patients who presented with hyperparathyroidism for occult pituitary or islet tumors could have lead to an overestimate of the frequency of positive tests among kindreds with isolated hyperparathyroidism. Comprehensive evaluation might have removed some of these kindreds from the hyperparathyroidism-only group and placed them in categories of pedigrees that have two or all three key MEN1-related neoplasms. However, our data are not inconsistent with previous reports of the frequency of MEN1 mutations in systematically evaluated pedigrees with familial hyperparathyroidism.39,40
The information presented here has value as a reflection of real-world experience. Although our data in some instances may have been limited, this is also true for referring physicians, who often receive incomplete family histories, and rarely have opportunities to systematically assess multiple family members within kindreds for the presence or absence of tumors in specific organs. The information we have been provided is that of our referring physicians and counselors.
References
Trump D, Farren B, Wooding C, Pang JT, Besser GM et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM 1996; 89: 653–669.
Gibril F, Schumann M, Pace A, Jensen RT . Multiple endocrine neoplasia type I and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine 2004; 83: 43–83.
Lairmore TC, Piersall LD, DeBenedetti KM, Dilley WG, Mutch M et al. Clinical genetic testing and early surgical intervention in patients with multiple endocrine neoplasia type 1 (MEN 1). Ann Surg 2004; 239: 637–647.
Skogseid B . Multiple endocrine neoplasia type 1. Br J Surg 2003; 90: 383–385.
Agarwal SK, Burns AL, Sukhodolets KE, Kennedy PA, Obungu VH et al. Molecular Pathology of the MEN1 Gene. Ann NY Acad Sci 2004; 1014: 189–198.
Doherty GM, Olson JA, Frisella MM, Lairmore TC, Wells SA, Norton JA . Lethality of Multiple Endocrine Neoplasia Type I. World J Surg 1998; 22: 581–587.
Marx S . Multiple Endocrine Neoplasia Type 1. In: Vogelstein B, Kinzler KW, eds. The Genetic Basis of Human Cancer. New York: McGraw Hill, 2002; 475–479.
Bassett JHD, Forbes SA, Pannett AAJ, Lloyd SE, Christie PT et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 1998; 62: 232–244.
Knudson A . Hereditary cancer: two hits revisited. J Cancer Res Clin Oncol 1996; 122: 135–140.
Friedman E, Sakaguchi K, Bale AE, Falchetti A, Streeten E et al. Clonality of parathyroid tumours in familial multiple endocrine neoplasia type 1. N Engl J Med 1989; 321: 213–218.
Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskold M . Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 1988; 332: 85–87.
Thakker RV, Bouloux P, Wooding C, Chotai K, Broad PM, Spurr NK et al. Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. N Engl J Med 1989; 321: 218–224.
Pannett AAJ, Thakker RV . Somatic mutations in MEN1 type 1 tumors, consistent with the knudson “two-hit” hypothesis. J Clin Endocrinol Metab 2001; 86: 4371–4374.
Busygina V, Suphapeetiporn K, Marek LR, Stowers RS, Xu T, Bale A . Hypermutability in a Drosophila model for multiple endocrine neoplasia type 1. Hum Mol Genet 2004; 13: 2399–2408.
Jin S, Mao H, Schnepp RW, Sykes SM, Silva AC, D'Andrea AD, Hua X . Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res 2003; 65: 4204–4210.
Chandrasekharappa SC, Guru SC, Manickam P, Olufemi S, Collins FS et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997; 276: 404–407.
The European Consortium on MEN1 Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. Hum Mol Genet 1997; 6: 1177–1183.
Pannett AAJ, Thakker RV . Multiple endocrine neoplasia type 1. Endocr Relat Cancer 1999; 6: 449–473.
Teh BT . Multiple endocrine neoplasia. J Intern Med 2003; 253: 588–589.
Giraud S, Zhang CX, Serova-Sinilnikova O, Wautot V, Salandre J et al. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet 1998; 63: 455–467.
Turner JJO, Leotlela PD, Pannett AAJ, Forbes SA, Bassett JH et al. Frequent Occurrence of an Intron 4 Mutation in Multiple Endocrine Neoplasia Type 1. J Clin Endocrinol Metab 2002; 87: 2688–2693.
Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P et al. Guidelines for Diagnosis and Therapy of MEN Type 1 and Type 2. J Clin Endocrinol Metab 2001; 86: 5658–5671.
Ciulla TA, Sklar RM, Hauser SL . A simple method for DNA purification from peripheral blood. Anal Biochem 1988; 174: 485–488.
Jeanpierre M . A rapid method for the purification of DNA from blood. Nucleic Acids Res 1987; 15: 9611.
Petty EM, Glynn M, Bale AE . Direct molecular diagnosis of multiple endocrine neoplasia type 1. Killeen AA, eds. Molecular Pathology Protocols. Totowa, NJ: Humana Press, 2000; 227–242.
Agarwal SK, Kester MB, Debelenko LV, Heppner C, Emmert-Buck MR et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet 1997; 7: 1169–1175.
Marx SJ, Nieman LK . Aggressive pituitary tumors in MEN1: Do they refute the two-hit model of tumorigenesis. J Clin Endocrinol Metab 2002; 87: 453–456.
Clerici T, Schmid C, Komminoth P, Lange J, Spinas GA, Brandle M . 10 Swiss kindreds with multiple endocrine neoplasia type 1: assessment of screening methods. Swiss Med Wkly 2001; 131: 381–386.
Kopp I, Bartsch D, Wild A, Schilling T, Nies C, Bergenfelz A et al. Predictive Genetic Screening and Clinical Findings in Multiple Endocrine Neoplasia Type I Families. World J Surg 2001; 25: 610–616.
Geerdink EAM, van der Luijt RB, Lips CJM . Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening?. Eur J Endocrinol 2003; 149: 577–582.
Skogseid B, Eriksson B, Gudmar L, Lorelius LE, Rastad J, Wide L et al. Multiple endocrine neoplasia type 1: A 10-year prospective screening study in four kindreds. J Clin Endocrinol Metab 1991; 73: 281–287.
Dean PG, van Heerden JA, Farley DR, Thompson GB, Grant CS, Harmsen WS et al. Are patients with multiple endocrine neoplasia type i prone to premature death?. World J Surg 2000; 24: 1437–1441.
Teh BT, Kytola S, Farnebo F, Bergman L, Wong FK et al. Mutation analysis of the MEN1 gene in multiple endocrine neoplasia type 1, familial acromegaly and familial isolated hyperparathyroidism. J Clin Endocrinol Metab 1998; 83: 2621–2626.
Mutch MG, Dilley WG, Sanjurjo F, DeBenedetti MK, Doherty GM, Wells SA Jr et al. Germline mutations in the multiple endocrine neoplasia type 1 gene: evidence for frequent splicing defects. Hum Mutat 1999; 13: 175–185.
Cavaco BM, Domingues R, Bacelar MC, Cardoso H, Barros L, Gomes L et al. Mutational analysis of Portuguese families with multiple endocrine neoplasia type 1 reveals large germline deletions. Clin Endocrinol 2002; 56: 465–473.
Human Gene Mutation Database. Available at: http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html. Accessed October 26, 2004.
Thakker RV . Multiple endocrine neoplasia type 1. Endocrinol Metab Clin 2000; 29: 541–567.
Tsukada T, Yamaguchi K, Kameya T . The MEN1 gene and associated diseases: An update. Endocr Pathol 2001; 12: 259–273.
Pannett AAJ, Kennedy AM, Turner JJO, Forbes SA, Cavaco BM, Bassett JHD et al. Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial isolated primary hyperparathyroidism. Clin Endocrinol 2003; 58: 639–646.
Warner J, Epstein M, Sweet A, Singh D, Burgess J, Stranks S et al. Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet 2004; 41: 155–160.
Acknowledgements
This work was supported by NIH grant R01 GM66079 to AEB and by T32 GM08753, which supported Drs. Klein and Salih. We especially thank Danielle Sullivan for her assistance with this project, as well as Daniel Dykas, the other members of the Bale Laboratory, and the many patients, physicians, and counselors who have entrusted our laboratory with their clinical MEN1 testing.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Klein, R., Salih, S., Bessoni, J. et al. Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med 7, 131–138 (2005). https://doi.org/10.1097/01.GIM.0000153663.62300.F8
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.GIM.0000153663.62300.F8
Keywords
This article is cited by
-
Long-term outcomes in MEN-1 patients with pancreatic neuroendocrine neoplasms: an Israeli specialist center experience
Endocrine (2020)
-
Late onset asymptomatic pancreatic neuroendocrine tumor – A case report on the phenotypic expansion for MEN1
Hereditary Cancer in Clinical Practice (2017)
-
Frequency of multiple endocrine neoplasia type 1 in a group of patients with pituitary adenoma: genetic study and familial screening
Pituitary (2014)
-
A novel menin gene deletional mutation in a little series of Italian patients affected by apparently sporadic multiple endocrine neoplasia type 1 syndrome
Journal of Endocrinological Investigation (2012)
-
An unusual association of neuroendocrine tumors in MEN 1A
Pituitary (2012)
