
Abstract
Purpose: It can be difficult to differentiate clinically between hemifacial microsomia (HFM) and Townes-Brocks syndrome (TBS). The distinction is important because TBS is inherited as an autosomal dominant trait, whereas HFM is sporadic.
Methods: We performed a retrospective analysis of eight patients with HFM-expanded spectrum and anal anomalies to determine whether this subset has TBS.
Results: Two patients had major phenotypic findings of TBS. Sequencing of SALL1, the gene mutated in TBS, in four of the eight patients revealed one with a C → T transition (resulting in a nonsense mutation R276X) at a previously identified mutational “hot spot.”
Conclusion: Patients with overlapping features of both syndromes should be screened for SALL1 mutations.
Similar content being viewed by others

Main
Hemifacial microsomia (HFM) and Townes-Brocks syndrome (TBS) have overlapping phenotypic features, and it can be difficult to differentiate between these two disorders.1,2 The classic triad of TBS includes anal anomalies (such as imperforate anus), hand and thumb anomalies, and auricular anomalies.3–6 Craniofacial abnormalities, including mandibular asymmetry, are rare in TBS. HFM with expanded spectrum (sometimes referred to as oculo-auricular-vertebral spectrum or “Goldenhar syndrome”) includes mandibular hypoplasia, cranial nerve weakness, auricular, vertebral, and ocular anomalies, most commonly epibulbar dermoids.7–9
TBS is caused by mutations in the SALL1 putative transcription factor.10 Several mutations in this gene have been found in both familial and sporadic cases of TBS.10–12 Similar phenotypes between TBS and HFM raise the possibility that some patients with HFM, particularly those with imperforate anus, may also have a SALL1 mutation.
It is essential to accurately diagnose TBS because it is inherited in an autosomal dominant fashion and, thus, has a 50% rate of transmission to offspring. HFM, on the other hand, is thought to be sporadic, and a 2–3% recurrence rate is generally quoted to families with an affected child.7,13,14
We studied eight patients who share some features of both TBS and HFM-expanded spectrum. Our goal was to determine if any of these patients had undiagnosed TBS. We analyzed the SALL1 gene in four of the eight patients by polymerase chain reaction (PCR) and direct sequencing, looking for mutations in the SALL1 gene. The possible presence of SALL1 mutations in this subset of patients is important because it would suggest that these overlapping clinical syndromes have a similar molecular pathophysiology.
MATERIALS AND METHODS
Patient selection and chart review
Based on a previous review of 121 patients with HFM-expanded spectrum,15 the medical records of five patients known to have anal abnormalities were analyzed in depth for possible other features of TBS. We reviewed records from clinical outpatient visits, inpatient hospitalizations and consultations, and earlier records obtained from other institutions. Two other patients were seen in the outpatient facility, and another was seen while hospitalized for an elective procedure during the course of this study, giving a total of eight. The patients' mandibular and auricular anomalies were graded by the OMENS system,8,15 where each letter of the acronym indicates one of the five major manifestations of HFM: orbit (O), mandible (M), ear (E), cranial nerves (N), and soft tissues (S). Each anatomic abnormality is assigned a number from 0 (normal) to 3 (most severe).
This study was approved by the Institutional Review Board at Children's Hospital, Boston. Informed consent was obtained from the four individuals who participated in the DNA sequencing portion of the study.
SALL1 mutational analysis
DNA was prepared from leukocytes of four patients (2, 3, 7, and 8) by standard methods. PCR was performed using 100–200 ng of genomic DNA, 10 pmol of each primer, 1× PCR buffer (10 mM Tris-HCl, pH 8.3, and 50 mM KCl, 1.5 mM MgCl2), 400 μM dNTPs, and 1 unit of Taq polymerase (Boehringer Mannheim). The PCR primers used (TF1, TR2, TF2.1, TR3, TR5, TF5, TR5.1) have been previously described.11 One additional primer corresponding with nucleotides 2491–2511 of the SALL1 gene (5′-GATGCTGCCCTCAGGACAGTC-3′) was used with the forward primer TF2.1. Two modified primers between nucleotides 546–1012 within exon 2 were used to focus on the “hot spot” area. Cycling conditions included an initial denaturation step at 94° × 4 minutes, followed by 35 cycles of 94° × 1 minute, 64° × 1 minute, 72° × 2 minutes, and a final extension step at 72° for 5 minutes. PCRs were performed in an MJ Research PTC-200 Pelter Thermal Cycler. PCR products were separated on standard agarose gels to check for amplification of the appropriately sized product, using both negative and positive PCR controls. PCR products were purified from the reaction mixture using Wizard PCR Prep Purification System (Promega). Purified double-stranded DNA templates were submitted to the DNA sequencing laboratory for direct double-stranded DNA sequencing using an ABI 373 automated sequencer with internal SALL1 primers.
RESULTS
Clinical summary of patients
Table 1 summarizes the clinical findings of the patients in this study. Of the eight patients, two were male, and the other six were female. All had facial asymmetry, structural auricular anomalies, and anal anomalies; thus, these findings are not listed in Table 1. None had a family history of similar deformities. Other findings were more variable. Three patients had extremity malformations. Four patients had ocular anomalies, with three of the four having epibulbar dermoids. Five of the eight patients had vertebral defects, and six of the eight patients had involvement of other organ systems.
SALL1 sequence data
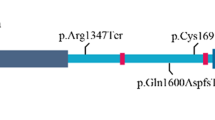
Exons 2 and 3 of the SALL1 gene, including intron/exon boundaries, were amplified by PCR and sequenced in four of the eight patients in our study (patients 2, 3, 7, and 8). Two patients declined to be tested, and two patients could not be located. All previously described mutations have been within exons 2 and 3, which contains all of the putative zinc finger motifs and encompasses 98% of the coding sequences of SALL1. We found a nonsense mutation at nucleotide 826/codon 276 (Arg → Ter, resulting from CGA → TGA) in one patient (patient 2) (Figs. 1 and 2). This C → T transition at a previously identified mutational “hot spot” is a de novo mutation in this family, since both parents did not carry the mutation, as determined by direct sequencing (Fig. 2). The remaining patients did not have any changes expected to be clinically significant in the area of the gene that was sequenced (data not shown). Exons 2 and 3 were fully sequenced in four normal controls as well.

Patient 2. Facial features: right type 1 mandibular hypoplasia; left type 2 microtia and right type 3 microtia; right facial nerve weakness. Extracraniofacial anomalies: bilateral duplicated thumbs; 1–2 syndactyly of the left foot; 4–5 syndactyly of the right foot with a cleft between first and second digits. Anogenital anomalies are hypospadias, a prominent perineal raphe, and anal tag.

DNA sequence chromatographs from patient 2, his parents, and a normal control. The sequence surrounding nucleotide 826 of the SALL1 gene is depicted, showing heterozygous C → T transition found in patient 2 (denoted by *) but not present in his parents and normal control.
DISCUSSION
Clinical differentiation between TBS and HFM-expanded spectrum can be confusing. TBS is characterized by the triad of anal, auricular, and hand anomalies.16 Anal abnormalities have been reported to occur in 95–97% of patients with TBS.3–6 Unilateral and bilateral auricular anomalies include “lop” or “satyr” ear, microtia, auricular pits, and tags,5 and sensorineural and/or conductive hearing loss.12 Renal anomalies are often seen, but less consistently so, and include hypoplastic or dysplastic kidneys, renal agenesis, multicystic kidneys, posterior urethral valves, and vesicoureteral reflux.6 Other associated anomalies of the genitourinary system are bifid scrotum and hypospadias.6 Vertebral defects have been described in only one TBS patient,17 and it has been proposed that this feature be used to differentiate TBS from VATER association or other overlapping conditions with vertebral anomalies.6 The occurrence of intrafamilial phenotypic variability in TBS has been well described.18,19
HFM is characterized by asymmetrical hypoplasia of the facial skeleton, ear, and facial soft tissues.9 Extracraniofacial anomalies occur in approximately 50% of patients.7,9 The term HFM “expanded spectrum” denotes extracraniofacial manifestations that are composed of malformations of the cardiac, skeletal, gastrointestinal, renal, and central nervous systems.15 There is significant variability in this condition, with the exception of facial asymmetry.8,9 Although there are no universally used diagnostic criteria, most studies include mandibular and auricular defects as obligatory features.9
Whereas TBS is an autosomal dominant disorder, many sporadic cases are described.6 HFM generally occurs sporadically, although cases of autosomal dominant inheritance have been reported.7,14 It is important to distinguish between these two diagnoses because of the reproductive risk associated with TBS. Several case reports have called attention to the phenotypic similarity between TBS and HFM. One report describes a three-generation family initially thought to have “Goldenhar-like” features. However, a child born in the third generation with imperforate anus raised the likely possibility of a diagnosis of TBS.2 Gabrielli et al.1 described a patient with microtia, preauricular tags, facial asymmetry, rib anomalies, and an anteriorly placed anus as a sporadic case of TBS versus “Goldenhar syndrome.” Moeschler and Clarren20 reported a mother and daughter with “hemifacial microsomia,” ear anomalies, and radial limb defects including duplicated triphalangeal thumbs. The daughter also had an anteriorly placed anus.
Our analysis found that only two of eight cases (patients 2 and 3) are likely have TBS. In addition to the typical features of TBS, patient 3 had an epibulbar dermoid, which is not characteristically seen in TBS, but does occur in 35% of patients with HFM.7 Patient 2 had no anal anomalies other than a prominent perineal raphe, but he did have other typical features of TBS, including microtia and hand anomalies. He also had cardiac, renal, and genital abnormalities, which occur in both TBS and HFM.6,7,15 However, his facial asymmetry was minor, with unilateral type 1 mandibular hypoplasia, and he did not have ocular or vertebral anomalies.
The gene for TBS codes for the SALL1 putative transcription factor and is located on chromosome 16q12.1. The SALL1 transcription factor shares homology to spalt (sal) of Drosophila melanogaster, where it is required for specification of anterior and posterior segmentation during development.21 The sal protein contains a highly conserved double zinc finger motif.10,21,22 The developmental expression pattern of sal homologs in mouse and Xenopus is consistent with the defects observed as a result of SALL1 gene disruption in patients with TBS. Human SALL1 is expressed in adult brain, kidney, and liver, and fetal brain.23
Mutations in the SALL1 gene have been found in both familial and sporadic cases of TBS.10–12,24 Kohlhase et al.10 initially reported mutations in one familial case and one sporadic case of TBS. A follow-up study analyzed 23 families for mutations in SALL1, and mutations were present in 9 families.11 In addition, a 1 base-pair deletion was found in a previously reported patient with overlapping features of TBS and HFM. In another recent report, mutations were identified in five familial and six sporadic cases of TBS, suggesting a mutational “hot spot” in sporadically occurring cases of TBS.12 In our study, one of the four patients whose SALL1 gene was sequenced (patient 2) showed a de novo nonsense mutation at nucleotide 826 (R276X) of the SALL1 gene, the putative “hot spot.” The R276X mutation would be predicted to result in a truncated protein and, thus, haploinsufficiency for SALL1 and was in the patient who had the least phenotypic overlap between TBS and HFM. Although functional studies might be helpful to ensure that this is the disease-causing mutation, the fact that an identical mutation has been described in six previous sporadic cases suggests that it is likely to be clinically important in this patient.
The other three patients in our study group did not have any changes in the SALL1 gene, other than likely polymorphisms. Specifically, there were no other changes detected at nucleotide 826, the proposed “hot spot.” This suggests that, in this subset of patients, there is no evidence of genetic overlap between these two syndromes. However, mutations in SALL1 have been identified in at least one other patient with features of these two syndromes.11 It is possible that our patients do have mutations outside of the sequenced region, such as in exon 1 or in an intron or flanking region of the gene. However, exon 1 contains only 2% of the coding sequences. A translocation breakpoint 180 kb away from the gene has been identified in one patient with TBS,12 suggesting that mutations in flanking regions of this gene can cause the TBS phenotype. Furthermore, large deletions or rearrangements could also be missed using only PCR and direct sequencing.
This is the seventh report of a mutation at nucleotide 826 of the SALL1 gene causing a sporadic case of TBS. The detection of this mutational “hot spot” has potential clinical applicability; for example, a simple and cost-effective diagnostic test for mutations surrounding nucleotide 826 could be developed to screen patients with features of TBS and no family history. DNA-based diagnostic testing for TBS is not yet clinically available. However, such a test would aid geneticists and genetic counselors in assisting families with a child who has features of TBS. If a mutation at nucleotide 826 is excluded, the remainder of the SALL1 gene could be sequenced easily. Although there is still likely to be diagnostic uncertainty in some patients with overlapping features of HFM and TBS, the development of DNA-based diagnostic testing would help some families or affected individuals who are interested in reproductive planning.
References
Gabrielli O, Bonifazi V, Offidani AM, Cellini A, Coppa GV, Giorgi PL . Description of a patient with difficult nosologic classification: Goldenhar syndrome or Townes-Brocks syndrome?. Minerva Pediatr 1993; 45: 459–462.
Johnson JP, Poskanzer LS, Sherman S . Three-generation family with resemblance to Townes-Brocks syndrome and Goldenhar/oculoauriculovertebral spectrum. Am J Med Genet 1996; 61: 134–139.
Ferraz FG, Nunes L, Ferraz ME, Sousa JP, Santos M, Carvalho C, Maroteaux P . Townes-Brocks syndrome: report of a case and review of the literature. Ann Genet 1989; 32: 120–123.
König R, Schick U, Fuchs S . Townes-Brocks syndrome. Eur J Pediatr 1990; 150: 100–103.
O'Callaghan M, Young ID . The Townes-Brocks syndrome. J Med Genet 1990; 27: 457–461.
Powell CM, Michaelis RC . Townes-Brocks syndrome. J Med Genet 1999; 36: 89–93.
Cohen MM, Rollnick BR, Kaye CI . Oculoauriculovertebral spectrum: an updated critique. Cleft Palate J 1989; 26: 276–286.
Vento AR, LaBrie RA, Mulliken JB . The O.M.E.N.S. classification of hemifacial microsomia. Cleft Palate Craniofac J 1991; 28: 68–76.
Cousley RRJ, Calvert ML . Current concepts in the understanding and management of hemifacial microsomia. Br J Plast Surg 1997; 50: 536–551.
Kohlhase J, Wischermann A, Reichenbach H, Froster U, Engel W . Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet 1998; 18: 81–83.
Kohlhase J, Taschner PEM, Burfeind P, Pasche B, Newman B, Blanck C, Breuning MH, ten Kate LP, Maaswinkel-Mooy P, Mitulla B, Seidel J, Kirkpatrick SJ, Pauli RM, Wargowski DS, Devriendt K, Proesmans W, Gabrielli O, Coppa GV, Wesby-van Swaay E, Trembath RC, Schinzel AA, Reardon W, Seemanova E, Engel W . Molecular analysis of SALL1 mutations in Townes-Brocks syndrome. Am J Hum Genet 1999; 64: 435–445.
Marlin S, Blanchard S, Slim R, Lacombe D, Denoyelle F, Alessandri J-L, Calzolari E, Drouin-Garraud V, Ferraz FG, Fourmaintraux A, Philip N, Toublanc J-E, Petit C . Townes-Brocks syndrome: detection of a SALL1 mutation hot spot and evidence for a position effect in one patient. Hum Mutat 1999; 14: 377–386.
Gorlin RJ, Cohen MM, Levin LS . Syndromes of the head and neck, 3rd ed. New York: Oxford University Press, 1990.
Stoll C, Viville B, Tressier A, Gasser B . A family with dominant oculoauriculovertebral spectrum. Am J Med Genet 1998; 78: 345–349.
Horgan JE, Padwa BL, LaBrie RA, Mulliken JB . OMENS-Plus: analysis of craniofacial and extracraniofacial anomalies in hemifacial microsomia. Cleft Palate Craniofac J 1995; 32: 405–412.
Townes PL, Brocks ER . Hereditary syndrome of imperforate anus with hand, foot, and ear anomalies. J Pediatr 1972; 81: 321–326.
Marlin S, Toublanc JE, Petit C . Two cases of Townes-Brocks syndrome with previously undescribed anomalies. Clin Dysmorphol 1998; 7: 295–298.
Monteiro de Pina-Neto J . Phenotypic variability in Townes-Brocks syndrome. Am J Med Genet 1984; 18: 147–152.
de Vries-Van der Weerd M-ACS, Willems PJ, Mandema HM, Kate LPT . A new family with the Townes-Brocks syndrome. Clin Genet 1988; 34: 195–200.
Moeschler J, Clarren SK . Familial occurrence of hemifacial microsomia with radial limb defects. Am J Med Genet 1982; 12: 371–375.
Kuhnlein RP, Frommer G, Friedrich M, Gonzalez-Gaitan M, Weber A, Wagner-Bernholz JF, Gehring WJ, Jackle H, Schuh R . spalt encodes an evolutionarily conserved zinc finger protein of novel structure which provides homeotic gene function in the head and tail region of the Drosophila embryo. EMBO J 1994; 13: 168–179.
Ott T, Kaestner KH, Monaghan AP, Schutz G . The mouse homolog of the region specific homeotic gene spalt of Drosophila is expressed in the developing nervous system and in mesoderm-derived structures. Mech Dev 1996; 56: 117–128.
Kohlhase J, Schuh R, Dowe G, Kühnlein RP, Jäckle H, Schroeder B, Schulz-Schaeffer W, Kretzschmar HA, Köhler A, Müller U, Raab-Vetter M, Burkhardt E, Engel W, Stick R . Isolation, characterization, and organ-specific expression of two novel human zinc finger genes related to the Drosophila gene spalt. Genomics 1996; 38: 291–298.
Salerno A, Kohlhase J, Kaplan BS . Townes-Brocks syndrome and renal dysplasia: a novel mutation in the SALL1 gene. Pediatr Nephrol 2000; 14: 25–28.
Acknowledgements
The authors are grateful to Va Lip for his technical assistance. This work was supported by grant NIHP30HD18655 in the Children's Hospital Mental Retardation Research Center.
Author information
Authors and Affiliations
Additional information
Partners Center for Human Genetics and Harvard Medical School, Boston, Massachusetts.
Rights and permissions
About this article
Cite this article
Keegan, C., Mulliken, J., Wu, BL. et al. Townes-Brocks syndrome versus expanded spectrum hemifacial microsomia: Review of eight patients and further evidence of a “hot spot” for mutation in the SALL1 gene. Genet Med 3, 310–313 (2001). https://doi.org/10.1097/00125817-200107000-00007
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/00125817-200107000-00007
Keywords
This article is cited by
-
Haploinsufficiency of BMP4 and OTX2 in the Foetus with an abnormal facial profile detected in the first trimester of pregnancy
Molecular Cytogenetics (2017)
-
Distinctive spine abnormalities in patients with Goldenhar syndrome: tomographic assessment
European Spine Journal (2015)
-
Oculoauriculovertebral spectrum with radial defects: a new syndrome or an extension of the oculoauriculovertebral spectrum? Report of fourteen Brazilian cases and review of the literature
European Journal of Human Genetics (2007)
