
Abstract
Lynch syndrome is the most common cause of inherited colorectal cancer, accounting for approximately 3% of all colorectal cancer cases in the United States. In 2009, an evidence-based review process conducted by the independent Evaluation of Genomic Applications in Practice and Prevention Working Group resulted in a recommendation to offer genetic testing for Lynch syndrome to all individuals with newly diagnosed colorectal cancer, with the intent of reducing morbidity and mortality in family members. To explore issues surrounding implementation of this recommendation, the Centers for Disease Control and Prevention convened a multidisciplinary working group meeting in September 2010. This article reviews background information regarding screening for Lynch syndrome and summarizes existing clinical paradigms, potential implementation strategies, and conclusions which emerged from the meeting. It was recognized that widespread implementation will present substantial challenges, and additional data from pilot studies will be needed. However, evidence of feasibility and population health benefits and the advantages of considering a public health approach were acknowledged. Lynch syndrome can potentially serve as a model to facilitate the development and implementation of population-level programs for evidence-based genomic medicine applications involving follow-up testing of at-risk relatives. Such endeavors will require multilevel and multidisciplinary approaches building on collaborative public health and clinical partnerships.
Genet Med 2012:14(1):152–162
Similar content being viewed by others

Main
Colorectal cancer (CRC) was diagnosed in approximately 143,000 individuals in the United States in 2007 (the most recent year US Cancer Statistics are available) and is the third most common cancer in men and women.1,2 CRC accounts for approximately 50,000 deaths each year and is largely preventable with recommended population screening.2,3 It is estimated that approximately 3% of CRCs are attributable to the hereditary condition Lynch syndrome (LS)–also referred to as hereditary nonpolyposis colorectal cancer.4,5,6 This autosomal dominant genetic disorder is associated with greatly increased risks for developing colorectal cancer and endometrial cancer.4,5,6,7, 8 Standard population CRC screening guidelines fail to provide early detection or prevention for most LS colon cancers as they tend to occur at young ages.4,5,6,7,8 The diagnosis of LS is primarily based on the presence of a germline mutation in a mismatch repair (MMR) gene and applies to both individuals with and without a cancer diagnosis.7 Additional details regarding the clinical and genetic characteristics of this condition and testing strategies are provided in the “Background” section.
Prior clinical studies have demonstrated the feasibility and efficacy of screening all cases of newly diagnosed CRC for LS, a process which has been referred to as “universal screening.”5,9,10,11,12 Furthermore, systematic evidence reviews have confirmed the analytic and clinical validity of available tumor screening tests to identify candidates for DNA analysis of the MMR genes associated with LS.7,8 These reviews also documented evidence of adequate test uptake among both individuals with CRC and their relatives, high levels of adherence to recommended surveillance, and minimal evidence of physical or psychological harms associated with testing or screening.6,7,8
Importantly, clinical research has found that of the 2.8% of subjects identified with LS by universal screening of all new CRCs, 50% were diagnosed over age 50 years, and 25% did not meet either the Amsterdam II diagnostic criteria13 or the revised Bethesda guidelines14 for LS.5 Thus, relying on age of diagnosis, family history, or tumor histology as defined by these criteria to determine who should undergo LS testing would miss a substantial number of affected individuals.15 Furthermore, clinical studies involving tumor screening of unselected endometrial cancers—the second most common malignancy associated with LS—have found a similar frequency (2.5%) of affected cases, many also not meeting age or family history criteria.16,17
In 2009, the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group (EWG) issued a recommendation regarding testing for LS in individuals with newly diagnosed CRC.6 The EWG is an independent, nonfederal panel involving multidisciplinary experts that uses a systematic evidence-based process for assessing the analytic validity, clinical validity, and clinical utility of genetic tests and other applications of genomic technology.18,19,20 Based on systematic reviews of the literature,7,8 the EWG determined that there is “sufficient evidence to recommend offering genetic testing for Lynch syndrome to individuals with newly diagnosed colorectal cancer in order to reduce morbidity and mortality in their relatives.”6 In addition, the EWG concluded that “there is moderate certainty that such a testing strategy would provide moderate population benefit.”6 The EWG did not directly address implementation issues nor did they find sufficient evidence to recommend a specific testing strategy among the four examined. However, a subsequent analysis suggested that tumor immunohistochemistry (IHC) for the MMR gene-protein products, followed by testing of the tumor for the V600E mutation in the BRAF gene (BRAF testing), is the most cost-effective approach.10
Given the EWG recommendation and the evidence for potential population health benefits of screening for LS in all individuals with newly diagnosed CRC (hereafter referred to as universal LS screening), the following Department of Health and Human Services Healthy People objective was submitted in 2009 by the Centers for Disease Control and Prevention (CDC) Office of Public Health Genomics (OPHG) and adopted as a 2020 developmental objective: “Increase the proportion of persons with newly diagnosed colorectal cancer who receive genetic testing to identify Lynch syndrome.”21 This objective is related to an overarching OPHG goal of reducing morbidity and mortality associated with LS in the United States through implementation of population-level genetic screening of new CRC cases and follow-up genetic testing of the relatives of patients with CRC found to carry a LS mutation (cascade testing).
To begin to construct the framework toward achieving this goal, OPHG convened a multidisciplinary meeting of clinical and public health experts, representing a variety of related specialties and organizations. The primary intent of this meeting was to explore possible approaches to population-level implementation of universal LS screening from the perspective of a public health/clinical partnership. Meeting participants included nongenetic physicians (gastroenterologists, surgeons, and family practitioners), genetics professionals (clinical geneticists, genetic counselors, and laboratory geneticists), epidemiologists and physicians from the CDC’s Division of Cancer Prevention and Control and the National Cancer Institute, and state level public health genetics professionals. The participants were provided with several relevant publications before the meeting, including the EWG recommendation and associated evidence reviews.6,7,8
The meeting sought to address the context of the environment in which the EWG recommendation can be implemented, with consideration of existing clinical paradigms and standards of care. The specific meeting objectives were to consider:
-
1
The opportunities and challenges surrounding implementation of the EWG recommendation regarding LS.
-
2
Strategies, tools, and infrastructure needed for implementation of universal LS screening and cascade testing on a national level.
-
3
Possible complementary approaches to maximize the identification of all individuals with LS in the US population.
This article provides background information relevant to understanding universal LS screening, summarizes the key discussion points of the meeting, and outlines multidisciplinary and multilevel strategies and approaches that were suggested to facilitate development of a population-level implementation protocol.
Background
LS fundamentals
LS is primarily caused by dominantly inherited mutations in the DNA MMR genes MLH1, MSH2, MSH6, and PMS2. It is characterized by a substantially increased risk for CRC (lifetime risk: 54–74% males and 30–52% females) with a mean age of onset ranging from 42 to 61.7,8,22,23,24,25,26 Additionally, females with LS have a 28–60% lifetime risk for endometrial cancer.23,24,25 LS is also associated with modest increased risks (generally 10% by age 70 years) for other cancers, among them gastric, ovarian, small bowel, urinary tract, pancreatic, and sebaceous gland tumors.27,28 It should be noted that cancer risks and age of onset vary dependent on which gene is involved, with lower risks and later age of onset noted for individuals with MSH6 and PMS2 mutations.26,29 Intensive cancer surveillance, including early initiation and frequent follow-up with colonoscopy, has been shown to reduce CRC incidence and related mortality in individuals with LS.6,30,31,32,33,34 Furthermore, evidence supports the efficacy of risk-reducing hysterectomy and salpingoophorectomy, and guidelines suggest offering this surgery as an option for women with LS.35,36,37 Current CRC screening recommendations include colonoscopy every 1–2 years beginning at age 20–25 years.36,37,38 Annual transvaginal ultrasound of the uterus and ovaries and endometrial sampling are also recommended, although efficacy remains to be documented as does the utility of screening for other LS cancers.6,36,37,38,39
Genetic evaluation for LS
Preliminary screening for LS can be performed on paraffin-embedded CRC tumor tissue, before pursuing more costly and complicated germline DNA mutation analysis for diagnostic purposes. Two types of tumor screening for LS are available: microsatellite instability (MSI) testing and IHC. It is estimated that up to 90–95% of LS tumors will exhibit high MSI in contrast to 10–20% of sporadic CRCs.7,40,41 However, lower levels of sensitivity are noted with use of fewer microsatellite markers (80–84%) and when germline MSH6 mutations are involved (77%).7 IHC can be used to determine expression of the MMR proteins MLH1, MSH2, MSH6, and PMS2 in the tumor tissue, with an estimated sensitivity of 83% (95% confidence interval: 75–89%) reported.7 In general, absence of protein expression is suggestive of a germline mutation in the related LS gene, allowing for targeted DNA mutation analysis. Specificity for both MSI and IHC is close to 90%.7 However, additional tumor testing (BRAF) is suggested if loss of MLH1 expression is found on IHC, as this occurs in 75% of cases as a result of somatic promoter hypermethlyation.7,42,43 In addition, there are other less common genetic explanations for loss of MLH1 or MSH2 expression without evidence of an associated MMR germline mutation.44,45 Because of this, if no MMR mutation is found, additional genetic testing may be warranted if early age of onset, multiple primary tumors, or a strong family history is present.
Preliminary screening with MSI and IHC provides concordant results in >94% of cases, although each method has its advantages and disadvantages.7,46 When tumor screening results are positive, DNA sequencing and deletion/duplication analysis of the indicated MMR gene(s) can be pursued, which will identify more than 90% of LS disease-causing mutations. Once a LS mutation is identified, at-risk relatives can be tested for the specific mutation in the family relatively inexpensively and with essentially 100% accuracy—an approach known as cascade testing or family tracing. Because LS is an autosomal dominant disorder, on average 50% of first-degree relatives will also be affected. Therefore, it is highly advantageous to systematically identify and test biologic relatives of people with LS by following a mutation through a family, testing those at 50% risk, and then, if positive, testing their children and other at-risk family members before symptoms would typically begin to appear. Those who do not carry the familial mutation are generally considered at average risk for CRC and other LS cancers and can typically undergo CRC screening according to standard population guidelines. In contrast, individuals who are mutation positive require intensive surveillance, which typically involves colonoscopy every 1–2 years beginning at age 25 years, and screening for extracolonic cancers and the option of preventive surgery.36
Potential population health impact
If LS genetic screening was routinely performed on all cases of CRC in the United States, approximately 4,200 affected individuals would be identified each year ( Figure 1 ). Assuming each individual identified with LS has four first-degree relatives with a 50% probability of inheriting the condition and eight second-degree relatives with a 25% probability, a total of 16,800 additional individuals with LS would be identified if all underwent evaluation for the familial mutation using a cascade testing approach. Based on conservative penetrance estimates, approximately half of these individuals (~8,400 cases) would develop CRC over the course of their lifetimes. Prospective studies of colonoscopic surveillance at 1–3-year intervals in individuals with LS have demonstrated a reduction in CRC incidence of 59–62%, indicating approximately 5,000 of these cancers would be prevented.31,32,47
Screening for LS on all newly diagnosed endometrial cancers (~42,000 new cases/year) would be expected to identify approximately 1,000 individuals with LS and 4,000 affected relatives, resulting in the potential to prevent an additional 1,500 CRC cases. Although LS screening of individuals with newly diagnosed endometrial cancer was not evaluated by the EWG, it is considered a logical next step based on available data and clinical application is already occurring.16
Although the aforementioned example is hypothetical and implies a best-case scenario, these figures illustrate that piloting implementation of universal LS screening on a population level is worth pursuing. This is supported by the EWG conclusion that benefits of testing for LS outweigh harms and the recent analysis which found universal LS screening to be highly cost-effective, even when more conservative uptake rates were applied.6,10
Existing application of routine genetic screening for LS
Several medical facilities across the country have existing protocols for routine LS genetic screening on newly diagnosed CRCs. Although no published statistics are available, a recent survey conducted on the National Society of Genetic Counselors cancer listserv reported that at least 30 centers/institutions are performing routine universal screening for LS, and several others are screening based on a specific criteria (e.g., age of diagnosis <50 years) (unpublished data). Some of these centers are also screening for LS on all or age subsets of newly diagnosed endometrial cancer cases. The protocols use various combinations of preliminary tumor testing (e.g., MSI only, IHC only, MSI and IHC, and IHC with BRAF) and differ with respect to consent and follow-up procedures.36 Systematic evaluation of these protocols and their effectiveness is needed to inform development of a population-level approach for universal LS screening, as existing data are limited. Notably, the one article published to date regarding clinical implementation of universal LS screening found lower uptake rates compared with those reported in research settings.48
Arguments against implementation of universal LS screening
It should be acknowledged that there have been arguments against the concept of universal LS screening. Hall49 contends that many aspects of the patient-level and social implications of adopting testing of all newly diagnosed CRC cases have not been adequately addressed. In addition to issues of access and cost associated with genetic counseling and testing, he noted the psychosocial burden to patients and families, limited societal benefit if there is poor uptake of testing and compliance with CRC screening, gaps in clinical resources and expertise, and the need to collect data regarding feasibility and effectiveness in real-world settings. Of these issues, lack of primary care provider knowledge and clinical expertise represents perhaps the most significant barriers to successful implementation of universal LS screening.
Peres11 interviewed clinicians from several institutions who noted difficulties with protocol logistics, concern that family history—already a challenge—would “fall by the wayside,” and the experience that many patients would “rather not know,” highlighting the need for at minimum, provision of information, and an “opt out” process rather than routine testing without patient knowledge. Ethical considerations regarding the level of consent required to perform LS screening on CRC tumors were recently addressed by Chubak et al.50 They suggested that although explicit informed consent should not be considered necessary for MSI testing, the issue is less certain for IHC testing given it can reveal specific genetic information regarding LS. They recommend further empirical study in this regard but emphasize that patients should be told results of both MSI and IHC screening, whether positive or negative.
In addition, there are issues regarding testing methodology which need to be addressed before a single universal LS screening protocol is promoted. For example, although preliminary tumor screening with IHC and BRAF has been suggested as the most cost-effective approach, this is likely related to improved specificity, rather than sensitivity. Furthermore, IHC scoring seems to show wide variability (kappa range: 0.49–0.79), suggesting that the diagnostic accuracy of current methods may be unacceptably low in some settings.51 Interpretation of the existing literature regarding the sensitivity and specificity of various tumor testing strategies is also confounded by incomplete testing of all four MMR genes and reports of ethnic variation.52 Further work is necessary to determine whether the limitations of IHC alone warrant performing both MSI and IHC or if these issues can be resolved by using centralized laboratories.
Finally, it is possible that the estimates of benefit in terms of reducing morbidity and mortality may not hold true for mutation- positive family members of LS cases detected through universal screening. Similar to the ascertainment bias which led to very high initial estimates of cancer risk in BRCA1/2 mutation carriers, the cancer penetrance and prevention rates typically quoted for LS are derived primarily from families identified due to multiple affected individuals and/or early age of onset.22,23,26,53 In addition, many studies have not included MSH6 and PMS2 mutation carriers, which are estimated to have lower penetrance and later age of onset for certain LS cancers compared with individuals with MLH1 or MSH2 mutations.26,29,54 The finding that many unselected CRC cases identified with LS do not fit classic diagnostic criteria raises the possibility that penetrance in these families may be lower and age of onset later, potentially resulting in a higher cost-benefit ratio if existing cancer screening protocols are applied. Hence, implementing universal LS screening should be accompanied by systematic collection of additional data to further assess clinical utility specifically in this context.
Meeting Summary
Following an introductory session and initial discussions, those meeting participants who had existing universal LS screening protocols or protocols in development shared their experiences, successes, and challenges, two of which are highlighted in this article. The Ohio State University (OSU) was one of the first centers to demonstrate the feasibility of screening all newly diagnosed CRC cases on both a research and a clinical basis, and their data provided part of the evidence for the EWG recommendation. The OSU current tumor testing protocol, which evolved from their previous research protocols, is illustrated in Figure 2 . Importantly, they found that the addition of BRAF mutation analysis substantially reduced the IHC screen-positive rate (8–10% vs. 20%) (unpublished data, used with permission). However, uptake of counseling and testing among individuals with positive tumor screens has been much lower with clinical implementation than the high levels observed in their research studies, where follow-up counseling and testing were free and travel was not required.9,48 Of 34 individuals deemed appropriate for genetic consultation—out of 270 who underwent screening in the first 2 years of the clinical program—only seven completed the testing process.48
The clinical protocol followed by the Huntsman Cancer Institute is illustrated in Figure 3 . To reduce the number of individuals who are “lost to follow up” and to improve testing uptake rates, their protocol automatically includes genetic counseling at the time of the postoperative appointment in patients whose tumor screens are positive. Their protocol currently excludes rectal cancers and colon cancers where surgery was not performed at their institute.
Identified successes and challenges
Multiple successes and challenges (both observed and potential) associated with the implementation of universal LS genetic screening and cascade testing of relatives were presented and discussed during the meeting. Table 1 provides a full list of these, whereas the most prominent themes which emerged in each category are highlighted below.
Successes:
-
1
Demonstrated effectiveness of LS screening on all newly diagnosed CRC cases versus relying solely on personal and/or family history.
-
2
Reduction in the number of cases requiring follow-up through the use of BRAF analysis.
Challenges:
-
1
Lack of provider knowledge of LS and testing issues.
-
2
Identification of the clinician with primary responsibility for reporting and following up on tumor screening results (e.g., pathologist, gastroenterologist, surgeon, genetic counselor, and oncologist) and coordination between specialists and primary care providers.
-
3
Design of effective strategies to ensure at-risk relatives of mutation-positive patients are identified and offered genetic counseling and testing.
Toward a public health approach to universal genetic screening for LS
Although there have been some individual institutional successes in implementing universal LS screening, for a population benefit to be realized, a broader coordinated approach is needed. Exploring challenges and successes involved in state newborn screening programs and population-level carrier screening for single gene conditions such as sickle cell disease and Tay Sachs disease will provide valuable information in this regard. Central to these is the need to ensure adequate education and training of all involved. In particular, those at the policy level will need to be convinced that as with newborn screening, although only a small percentage of individuals are actually found to be affected, the impact of the diagnosis on health outcomes is profound, particularly as it applies to the multiple family members likely to be at risk for LS.
As with carrier and newborn screening programs, developing implementation protocols for universal LS screening will require significant collaborations between healthcare systems and public health agencies, with strategies and interventions targeted at multiple levels across the healthcare continuum. A framework for consideration of a multilevel approach is that of Taplin.55,56 This framework places the individual patient at the core, surrounded by concentric layers of key healthcare influences building on each other: family and social supports, the provider/healthcare team, the organization/practice setting, the local community environment, the state health policy environment, and the national health policy environment. Similarly, as outlined by Khoury,57 successful implementation and dissemination of evidence-based genomic medicine will require conscious consideration and integration of multiple components, including clinical and laboratory perspectives; behavioral, social, and communication factors; healthcare organizations; public health system perspectives; and policy, oversight, and regulatory frameworks.
To begin conceptualizing a multilayered approach toward universal LS screening on a population level, meeting participants considered the framework described earlier and worked in small groups to identify (1) the primary “layers” of importance; (2) who should be targeted in each layer—including what do they need and how do we reach them; (3) mechanisms to integrate the layers; and (4) ways to monitor uptake and outcomes of implementation. Both divergent and common themes were identified as part of this activity and are reviewed in the following paragraphs.
Considering an “outside-in” approach, one proposal was to convene national-level (e.g., National Institutes of Health) conferences to build consensus on (1) practical issues such as conveying test results to patients and family members; (2) development of standards and guidelines for pathology reports requiring inclusion of MSI/IHC results; (3) reviewing and revising practice recommendations through involvement of professional societies; and (4) identifying and addressing inadequacies in policy, facilities, and the workforce. State agencies could require LS test results to be included in tumor registries as a means of monitoring implementation. Genetic workforce issues could potentially be addressed at the state level by supporting a few regional genetic counselors to provide test follow-up. Buy in from payers is considered critical and could be spearheaded by the Centers for Medicare and Medicaid Services to cover not just the tumor screening, but follow-up genetic consultation, testing, and surveillance for individuals found to have LS and their at-risk family members. Involvement of hospital/medical group practices, primary care providers, and multiple provider specialties (e.g., pathology, gastroenterology, surgery, and genetics) will be essential to inform and support development of guidelines by national organizations, agencies, and professional societies.
Another focus was “multi-level education,” ensuring entities at all layers—patients, family members, healthcare providers, public and private health systems, policy makers—are operating from the same understanding of the rationale for universal LS screening, the importance of genetic counseling and diagnostic testing for individuals whose tumor screens are positive, and the need to follow through with identification of at-risk family members and ensure appropriate surveillance of mutation-positive individuals. Public health entities can contribute on a state and national level by providing fact sheets and toll-free numbers for individuals and families, as well as centralized access to information for providers and payers. Professional guidelines, electronic medical record reminders, and clinical decision support tools could be used to enhance provider education and facilitate practice change. Providers will need access to an information technology infrastructure to facilitate this knowledge transfer, as they will be essential to educating individual patients and their family members. Creation of regionalized guidelines for healthcare systems, a national registry providing a network to connect families and providers, and ensuring compliance and quality assurance through tumor registries and central pathology groups were additional solutions offered to facilitate integration and monitoring. In particular, requiring LS tumor screening results to be included on all CRC pathology reports would allow for compliance to be monitored through the tumor registry system and provide an avenue to identify patients where appropriate follow-up needs to be confirmed. However, the time lapse between pathologic diagnosis and inclusion in the tumor registry and issues of patient confidentiality may limit what can be achieved through this method.
A third major approach concentrated on using the concepts and processes involved in newborn screening as a model for implementing universal LS screening on a population level. The original conceptualization of this idea is attributed to Heather Hampel and Albert de la Chapelle of the OSU (personal communication). National and/or state policy recommendations for universal LS screening would be needed, along with the creation of centralized state or regional laboratories. Individual pathology laboratories would be required to send a core of every CRC tumor specimen to the central laboratory, which would be set up to perform IHC on multiple samples simultaneously using tissue microarray technology. This should reduce cost and reading time per specimen and help to mitigate concerns regarding variability of IHC testing. A reliable reporting system would be essential, preferably by electronic medical record, as would the monitoring of appropriate follow-up. Results could be linked to state cancer registries as a check/balance to ensure the system is working. State level genetics professionals could be designated to facilitate follow-up counseling and testing of patients with positive tumor screens, particularly for systems where there is no easy access to genetic services. These professionals could network with their counterparts in other states to coordinate communication and follow-up with family members. Integration of the layers could be facilitated by providing incentives tied to outcomes both for the individual practitioners and for healthcare systems. These activities could also help improve understanding of CRC disparities in the US population.
Additional common themes and considerations emerged from the meeting discussions. It was recognized that pathologists and their related national organizations (e.g., the College of American Pathologists and the Association for Molecular Pathology) will be critical to the process of universal LS screening. Parallel to this will be the need for centralized/regional laboratories that will address issues of standardization of results and reporting, as well as quality control. Participants in general believed that the evidence is also compelling to consider universal genetic screening for LS in all cases of newly diagnosed endometrial cancer and that adding this and other Lynch-specific cancers (e.g., sebaceous gland tumors) once protocols are in place for CRC should be relatively straightforward.
It was acknowledged that recognizing and emphasizing the direct benefit to the patient with CRC diagnosed with LS will assist in addressing challenges associated with informed consent and insurance coverage. Although the EWG concluded at the time of their publication that insufficient evidence existed to alter treatment of LS-related CRCs, two systematic reviews have reported a potential adjuvant treatment impact of knowing the MSI status of CRC.58,59 Specifically, MSI-H colon cancers have shown a better prognosis and differential response to chemotherapy—particularly reduced efficacy of 5-fluorouracil.58,59 A recent retrospective cohort analysis also demonstrated a substantially lower risk for metachronous CRC in individuals with LS undergoing extensive versus segmental colon resection.60 In addition, the increased risk for developing second primary tumors (metachronous CRC and other cancers) will impact the screening and management of patients with CRC who are found to have LS. For example, standard practice after CRC diagnosis and treatment is to repeat colonoscopy at 1-year, then 3-year, and subsequently at 5-year intervals, assuming no abnormalities are noted.61 In contrast, ongoing colonoscopy every 1–2 years is standard of care for individuals with a LS-related CRC.6,36,37,38,62 Furthermore, screening with transvaginal ultrasound/endometrial biopsy and consideration of risk-reducing hysterectomy and oophorectomy are recommended for women with LS and a previous CRC.36,37,38,62
It was also noted that in accordance with the EWG recommendation, although family history should not be used to exclude individuals with newly diagnosed CRC from LS screening, it remains a valuable and important tool to identify unaffected individuals in the general population who may benefit from genetic evaluation for hereditary CRC. Thus, education of consumers and providers regarding the importance of family history and features suggestive of LS should continue to be encouraged. With increasing focus on the patient-centered medical home model, primary care providers will play an increasing role in identifying persons at high risk and referring patients for genetic counseling and predictive genetic testing.48 However, attempting to initiate a complementary and concurrent universal screening approach to identify healthy individuals at risk for LS based on family history was not considered feasible at this time.
The particular challenges of implementing universal LS genetic screening within the fragmented US healthcare system were discussed, including the need to enhance recognition of and avenues for addressing familial implications of the disease; dealing with the complexities of different insurance plans and limited avenues for reimbursement; and ensuring testing and surveillance of individuals identified with LS who may not have a primary care provider. Such issues as these may be most effectively addressed by public health entities partnering with those involved in clinical care, as well as with payers and policy makers. Using existing cancer registries and public health surveillance protocols could be one potential way to address some of these integration challenges.
Finally, it was recognized that working toward national implementation of screening for LS on all newly diagnosed CRC cases will require pilot studies and demonstration projects on a smaller scale, involving various types of healthcare systems and different state or regional platforms. Prospective data collection and comparative effectiveness research should be used to evaluate the benefits and limitations of implementing a newborn screening-like approach to universal LS screening in contrast to other potential or existing service delivery models. Further research on “real-world” clinical implementation is needed to collect additional data regarding uptake of genetic counseling and molecular testing of screen-positive patients, as well as the initiation of cascade testing and appropriate cancer surveillance in relatives of those individuals identified with LS. Such data will be critical to justify the cost and infrastructure requirements associated with initiating universal LS screening on a national level. It was further noted that in this era of limited resources, careful consideration of the appropriateness of using public health dollars toward population-level universal LS screening is warranted.
Meeting conclusions and recommendations
The following seven points summarize the key conclusions and recommendations that emerged from the meeting:
-
1
Genetic screening of all newly diagnosed CRC cases for LS (universal LS screening) can theoretically result in population health benefits, and feasibility has been demonstrated in research and clinical settings.
-
2
Using a public health approach strongly integrated with all aspects of clinical care may provide the greatest opportunity for successful implementation on a regional or national scale.
-
3
There are several challenges and barriers to implementation of universal LS screening that need to be evaluated and addressed before consideration of large-scale efforts at the state, regional, or national level.
-
4
Education of clinicians, patients, families, healthcare system administrators, payers, and state and national public health entities and policy makers will be critical to any national effort.
-
5
National level conferences should be convened to allow further dialog among key organizations, groups, and individuals regarding development of protocols, policies, and guidelines addressing universal LS screening on a state and/or national level.
-
6
Serious consideration should be given to the paradigm of newborn screening as a model for implementing universal LS screening on a national level.
-
7
Carefully constructed pilot implementation projects and “real-world” studies are needed to demonstrate effectiveness and provide additional evidence of the feasibility and utility of population-level universal LS screening.
Summary
Despite the explosion over the past decade in the number and availability of genetic tests for a variety of human conditions and diseases, there have been no significant efforts on a national level to facilitate widespread genetic screening to prevent adult onset conditions in relatives of affected individuals using a public health model. In particular, although newborn screening protocols, which have existed in some form since the late 1960s, have continued to add rare genetic diseases to their panels, universal genetic screening for other health conditions at different life stages has not been attempted. LS in many respects may be the ideal genetic condition to launch this new era in public health genomics. This is because the condition—although rare in the general population—accounts for 2–3% of both CRC and endometrial cancers; relatively inexpensive and accurate preliminary screening of tumor tissue is available; sensitive and specific molecular genetic testing can facilitate identification of mutation-positive individuals and allow for cascade testing in relatives; identification of the mutation status of family members can reduce morbidity and mortality by initiating appropriate cancer screening for those at risk and avoiding unnecessary and costly screening of those who are mutation negative; and evidence exists of the potential impact of the diagnosis of LS on treatment, subsequent management, and surveillance for additional cancers in individuals with CRC.
At the same time, there are many questions which need to be answered before the EWG recommendation regarding universal LS screening can be implemented on a state, regional, or nationwide population level. Central to these issues is the need to educate consumers, providers, healthcare system administrators, payers, public health agencies, and state/federal policy makers regarding LS and its familial implications. There is a need for pilot population studies to gather more data, including but not limited to demonstration of the following: high levels of compliance with tumor tissue screening across multiple institutions; success of protocols designed to ensure accurate and timely reporting and follow-up of positive tumor results; reasonable levels of genetic counseling/testing uptake by patients with CRC with a positive tumor screen and family members of mutation-positive individuals; compliance with recommended cancer screening protocols and management; benefit to both the patient and family members; and minimal evidence of psychosocial and societal harm. Although many of the aforementioned issues have been demonstrated in high-risk clinic and research settings, clinical application on a wide-spread population level requires more study in these areas. These data will be essential to justify the expenditure on infrastructure that will be required to make population-level universal screening for LS a reality. It is only through multidisciplinary, multistakeholder involvement and cooperation—ideally facilitated though clinical/public health partnerships—that such accomplishments will be attainable.
Disclosure
Heather Hampel, MS, has received an honorarium from Myriad Genetic Laboratories, Inc. to serve on a Lynch syndrome Advisory Panel. The other authors declare no conflict of interest.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention or the National Cancer Institute.

Potential population health impact of universal screening for Lynch syndrome. CRC, colorectal cancer; EC, endometrial cancer; LS, Lynch syndrome.

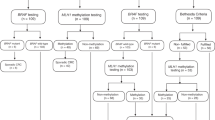
The Ohio State University—clinical Lynch syndrome screening strategy: follow-up of IHC testing on all colorectal cancers. IHC, immunohistochemistry; MSI, microsatellite instability. aReferral to genetic counseling if warranted by clinical or family history.

Huntsman Cancer Institute—routine testing strategy for Lynch syndrome. IHC, immunohistochemistry. aPatients usually seen during surgical follow-up appointment.
References
U.S. Cancer Statistics Working Group. United States cancer statistics: 1999–2007 incidence and mortality web-based report. Department of Health and Human Services, Centers for Disease Control and Prevention, and National Cancer Institute: Atlanta, GA, 2010. http://www.cdc.gov/uscs. Accessed 15 January 2011.
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ . Cancer statistics, 2009. CA Cancer J Clin 2009;59:225–249.
Winawer S, Fletcher R, Rex D, et al. Colorectal cancer screening and surveillance: clinical guidelines and rationale-Update based on new evidence. Gastroenterology 2003;124:544–560.
Lynch HT, de la Chapelle A . Hereditary colorectal cancer. N Engl J Med 2003;348:919–932.
Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352: 1851–1860.
Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009;11:35–41.
Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN . EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med 2009;11:42–65.
AHRQ. Hereditary nonpolyposis colorectal cancer: diagnostic strategies and their implications, 2007. http://www.ahrq.gov/clinic/tp/hnpcctp.htm. Accessed 15 December 2010.
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008;26: 5783–5788.
Mvundura M, Grosse SD, Hampel H, Palomaki GE . The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med 2010;12:93–104.
Peres J . To screen or not to screen for Lynch syndrome. J Natl Cancer Inst 2010;102:1382–1384.
Hampel H . Point: justification for Lynch syndrome screening among all patients with newly diagnosed colorectal cancer. J Natl Compr Canc Netw 2010;8:597–601.
Vasen HF, Watson P, Mecklin JP, Lynch HT . New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999;116:1453–1456.
Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261–268.
de la Chapelle A, Palomaki G, Hampel H . Identifying Lynch syndrome. Int J Cancer 2009;125:1492–1493.
Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 2006;66:7810–7817.
Hampel H, Panescu J, Lockman J, et al. Comment on: Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) among Endometrial Cancer Patients. Cancer Res 2007;67:9603.
Evaluation of Genomic Applications in Practice and Prevention. http://www.egappreviews.org. Accessed 15 December 2010.
Teutsch SM, Bradley LA, Palomaki GE, et al. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group. Genet Med 2009;11:3–14.
Botkin JR, Teutsch SM, Kaye CI, et al. Outcomes of interest in evidence-based evaluations of genetic tests. Genet Med 2010;12:228–235.
Healthy People 2020: genomics objectives. http://www.healthypeople.gov/2020/topicsobjectives2020/objectiveslist.aspx?topicid=15. Accessed 15 December 2010.
Barrow E, Alduaij W, Robinson L, et al. Colorectal cancer in HNPCC: cumulative lifetime incidence, survival and tumour distribution. A report of 121 families with proven mutations. Clin Genet 2008;74:233–242.
Dunlop MG, Farrington SM, Carothers AD, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 1997;6:105–110.
Hampel H, Stephens JA, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology 2005;129:415–421.
Stoffel E, Mukherjee B, Raymond VM, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology 2009;137:1621–1627.
Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011;305:2304–2310.
Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009;75:141–149.
Watson P, Vasen HF, Mecklin JP, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer 2008;123: 444–449.
Senter L, Clendenning M, Sotamaa K, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 2008;135:419–428.
Vasen HF, Abdirahman M, Brohet R, et al. One to 2-year surveillance intervals reduce risk of colorectal cancer in families with Lynch syndrome. Gastroenterology 2010;138:2300–2306.
Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000;118:829–834.
Järvinen HJ, Renkonen-Sinisalo L, Aktán-Collán K, Peltomäki P, Aaltonen LA, Mecklin JP . Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation-negative family members. J Clin Oncol 2009;27:4793–4797.
de Vos tot Nederveen Cappel WH, Nagengast FM, Griffioen G, et al. Surveillance for hereditary nonpolyposis colorectal cancer: a long-term study on 114 families. Dis Colon Rectum 2002;45:1588–1594.
de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006;130:665–671.
Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med 2006;354:261–269.
National Comprehensive Cancer Network. Clinical practice guidelines in oncology: colorectal cancer screening (v1.2010), 2010. http://www.nccn.org/professionals/physician_gls/PDF/colorectal_screening.pdf. Accessed 15 December 2010.
Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006;296:1507–1517.
Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007;44:353–362.
Koornstra JJ, Mourits MJ, Sijmons RH, Leliveld AM, Hollema H, Kleibeuker JH . Management of extracolonic tumours in patients with Lynch syndrome. Lancet Oncol 2009;10:400–408.
Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998;338:1481–1487.
Giardiello FM, Brensinger JD, Petersen GM . AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterology 2001;121:198–213.
Menigatti M, Di Gregorio C, Borghi F, et al. Methylation pattern of different regions of the MLH1 promoter and silencing of gene expression in hereditary and sporadic colorectal cancer. Genes Chromosomes Cancer 2001;31:357–361.
Capel E, Fléjou JF, Hamelin R . Assessment of MLH1 promoter methylation in relation to gene expression requires specific analysis. Oncogene 2007;26:7596–7600.
Hitchins MP, Ward RL . Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J Med Genet 2009;46:793–802.
Niessen RC, Hofstra RM, Westers H, et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer 2009;48:737–744.
Baudhuin LM, Burgart LJ, Leontovich O, Thibodeau SN . Use of microsatellite instability and immunohistochemistry testing for the identification of individuals at risk for Lynch syndrome. Fam Cancer 2005;4:255–265.
Stupart DA, Goldberg PA, Algar U, Ramesar R . Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Colorectal Dis 2009;11:126–130.
South CD, Yearsley M, Martin E, Arnold M, Frankel W, Hampel H . Immunohistochemistry staining for the mismatch repair proteins in the clinical care of patients with colorectal cancer. Genet Med 2009;11:812–817.
Hall MJ . Counterpoint: implementing population genetic screening for Lynch Syndrome among newly diagnosed colorectal cancer patients–will the ends justify the means? J Natl Compr Canc Netw 2010;8: 606–611.
Chubak B, Heald B, Sharp RR . Informed consent to microsatellite instability and immunohistochemistry screening for Lynch syndrome. Genet Med 2011;13:356–360.
Overbeek LI, Ligtenberg MJ, Willems RW, et al. Interpretation of immunohistochemistry for mismatch repair proteins is only reliable in a specialized setting. Am J Surg Pathol 2008;32:1246–1251.
Salovaara R, Loukola A, Kristo P, et al. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000;18:2193–2200.
Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999;81:214–218.
Baglietto L, Lindor NM, Dowty JG, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst 2010;102:193–201.
Taplin SH, Clauser S, Rodgers AB, Breslau E, Rayson D . Interfaces across the cancer continuum offer opportunities to improve the process of care. J Natl Cancer Inst Monographs 2010;2010:104–110.
Taplin SH. Talk presented at Multilevel interventions across the cancer care continuum. Mutilevel interventions in health care: building the foundation for future research. Las Vegas, Nevada, 2011. http://dccps.nci.nih.gov/mli/background-2011.pdf. Accessed 10 June 2011.
Khoury MJ, Bowen MS, Burke W, et al. Current priorities for public health practice in addressing the role of human genomics in improving population health. Am J Prev Med 2011;40:486–493.
Popat S, Hubner R, Houlston RS . Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–618.
Des Guetz G, Schischmanoff O, Nicolas P, Perret GY, Morere JF, Uzzan B . Does microsatellite instability predict the efficacy of adjuvant chemotherapy in colorectal cancer? A systematic review with meta-analysis. Eur J Cancer 2009;45:1890–1896.
Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60:950–957.
Rex DK, Kahi CJ, Levin B, et al. Guidelines for colonoscopy surveillance after cancer resection: a consensus update by the American Cancer Society and the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2006;130:1865–1871.
American Gastroenterological Association medical position statement: hereditary colorectal cancer and genetic testing. Gastroenterology 2001;121:195–197.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bellcross, C., Bedrosian, S., Daniels, E. et al. Implementing screening for Lynch syndrome among patients with newly diagnosed colorectal cancer: summary of a public health/clinical collaborative meeting. Genet Med 14, 152–162 (2012). https://doi.org/10.1038/gim.0b013e31823375ea
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.0b013e31823375ea
Keywords
This article is cited by
-
Novel strategy of multiple-locus variable number tandem repeats analysis for genetic fingerprinting of human
Genes & Genomics (2023)
-
Universal tumor screening for Lynch syndrome: perspectives of Canadian pathologists and genetic counselors
Journal of Community Genetics (2019)
-
Effective Identification of Lynch Syndrome in Gastroenterology Practice
Current Treatment Options in Gastroenterology (2019)
-
Choosing not to undergo predictive genetic testing for hereditary colorectal cancer syndromes: expanding our understanding of decliners and declining
Journal of Behavioral Medicine (2017)
-
Preferences for genetic testing for colorectal cancer within a population-based screening program: a discrete choice experiment
European Journal of Human Genetics (2016)
