
Abstract
Objectives
To investigate retinal microstructure of patients affected with malattia leventinese (MLVT) and mutation in the EFEMP1 gene using high-resolution optical coherence tomography (OCT).
Methods
Patients diagnosed with MLVT received a comprehensive eye exam, full-field and multifocal electroretinogram testing and imaging with a high-resolution Fourier domain OCT (Fd-OCT, UC Davis Medical Center, Davis, USA; axial resolution: 4.5 μm, acquisition speed: 9 frames s−1, 1000 A scans s−1) combined with a flexible scanning head (Bioptigen Inc. Durham, NC, USA).
Results
Two related patients aged 30 and 60 years, with MLVT and identified c.R345W mutation in the EFEMP1 gene, were tested. Mother and daughter showed a variable phenotype with reduced vision function in the younger patient, whereas the mother had a ‘form frustre’. Fd-OCT revealed extensive or focal sub-retinal pigment epithelium (RPE) deposits, separation of RPE and Bruch's membrane, and disruption of the photoreceptor outer and inner segment layers. No outer retinal changes were visible outside areas with sub-RPE deposits.
Conclusion
Retinal structure in EFEMP1 retinal dystrophy is reflected by morphological changes within the RPE/Bruch's membrane complex with accumulation of sub-RPE material associated with disrupted photoreceptor integrity. The pattern of microstructural retinal abnormalities is similar but with a different extent in patients with variable phenotypes.
Similar content being viewed by others
Introduction
A subset of genetically related autosomal dominant drusen (OMIM 126600) were originally described as Doyne honeycomb retinal dystrophy in Britain and as malattia leventinese (MLVT) in Switzerland, which are caused by a single missense mutation Arg345Trp in the EFEMP1 gene.1 Characteristic signs are early onset drusenoid deposits at the posterior pole and in the peripapillary area with a possible radial distribution, increasing confluence with age and inter- and intrafamilar variability.2 Fu et al.3 suggested that mutant Efmp1 protein might change the extracellular matrix in Bruch's membrane resulting in basal laminar deposits. Retinal layer characterization, using time-domain optical coherence tomography (OCT), has demonstrated diffuse alterations of retinal pigment epithelium (RPE)/Bruch's membrane complex4, 5 with preservation of the neurosensory layers.5 Detailed analyses, in particular of the outer retinal layers have, however, been limited by the image resolution of commercial time-domain instrumentation. Here, we demonstrated significant photoreceptor changes in two cases with variable EFEMP1 associated retinal dystrophy.
Methods
Two patients diagnosed with the MLVT phenotype and identified c. R345W mutation in the EFEMP1 gene (Carver laboratory, Iowa, IA, USA) were recruited through the Ocular Genetics Clinic at the Hospital for Sick Children in Toronto, Canada. Written informed consent was obtained from patients. The project was approved by the Research Ethics Board at Sick Kids as well as the University of California, Davis Institutional Review Board, and conducted in accordance with the Tenets of Helsinki. We certify that all applicable institutional and governmental regulations concerning the ethical use of human volunteers were followed during this research.
Vision function assessment included best-corrected monocular near and distance visual acuity, colour vision and contrast sensitivity. Retinal responses were recorded using full-field electroretinography (ffERG; International Society for Clinical Electrophysiology of Vision (ISCEV) standard)6 and multifocal electroretinography (mfERG; 103 hexagons, ISCEV recommendation).
Retinal image acquisition was achieved using a custom built high-speed, high-resolution Fd-OCT system7 (axial resolution: 4.5 μm; acquisition speed: 9 frames −1s, 1000 A scans s−1) constructed at UC Davis with a sample arm scanning head (Bioptigen Inc.). Horizontal scans of 6 mm length or a volumetric scan series permitting image acquisition over an area of 6 × 6 mm and OCT fundus reconstruction were registered through the macular area. Raw image data were post-processed and retinal layers were identified as described earlier (Figure 1).8, 9

Six-millimetre horizontal Fd-OCT scan through the right macula of a 36-year-old control. CL, connecting cilia; GCL, ganglion cell layer; ILM/NFL, internal limiting membrane/nerve fiber layer; INL, inner nuclear layer; IPL, inner plexiform layer; ISL, inner segment layer; OLM, outer limiting membrane; ONL, outer nuclear layer; OPL, outer plexiform layer; OSL, outer segment layer; RPE/BM, retinal pigment epithelium/Bruch's membrane; VM, Verhoeff's membrane.
Results
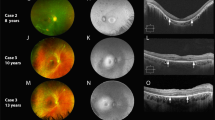
A 33-year-old woman (case 1) and her 60-year-old asymptomatic mother (case 2) of French/Scottish/Dutch ancestry were assessed. Results of vision function are summarised in Table 1. Fundus examination showed small, round, yellowish-white deposits throughout the macular and peripapillar area with perifoveal pigmentary changes and RPE atrophy (case 1) (Figure 2). The mother's fundus revealed two areas of extrafoveal confluent white deposits with central RPE atrophy and few retinal deposits in the nasal fundus in both eyes. (Figure 3).

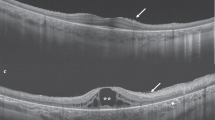
Composite of fundus photograph and serial horizontal Fd-OCT scans (6 mm) of the right and left eyes of patient no.1. The locations of OCT B-scans are denoted by lines (a–e) shown on the OCT fundus image (intensity projection of the OCT volume), which is superposed and registered to the colour fundus photo. Lower panel shows a virtual C-scan at the level of the photoreceptor inner/outer segment junction segmented from the reconstructed OCT volume. B-scans illustrate extensive deposits (denoted by a star in one scan) in the sub-RPE area with separation between the RPE and Bruch's membrane (arrow denotes the location in two scans). Circles provide example of disruption of the outer segment layer, inner segment layer, and outer nuclear layer.

Composite of colour fundus photograph and serial horizontal Fd-OCT scans (6 mm) and virtual C-scan (for description see Figure 2) of the right and left eye of patient no.2. The locations of OCT B-scans are denoted by lines (a–e) shown on the OCT fundus image. Circels denote areas of sub-RPE deposits with corresponding photoreceptor disruption. B-scans illustrate disruption of the outer retinal layers (star denotes area in three scans). Circles provide example of cone-shaped deposits in the RPE area extending into inner retinal layers. Subfoveal photoreceptor layers were preserved.
Macular scans from case 1 demonstrated a separation of Bruch's membrane and RPE by extensive dense highly reflective material in the sub-RPE zone (Figure 2). It appeared that those deposits are pushing and elevating the outer retinal layers. Outer segment layer, inner segment layer, and outer nuclear layer were disrupted more in the subfoveal than in the extrafoveal areas. Outer limiting membrane structure was disturbed. Retinal microstructure showed similar but more focal changes in correspondence to the visible fundus abnormalities with preserved subfoveal layers in case 2 (Figure 3). Photoreceptor disruption was confined to areas with sub-RPE deposits in both cases.
Discussion
High-resolution retinal imaging permitted identification of disrupted photoreceptor layers in two patients with variable MLVT phenotype caused by the c. R345W mutation in the EFEMP1 gene for the first time. Our findings are in agreement with histopathological studies of MLVT cases, which demonstrated formation of hyaline substances between retina and choroid with extension through the outer limiting membrane in some cases. There, destroyed rod and cone photoreceptor layers and outer nuclear layer but unchanged inner retina layers were described.10, 11
Efemp1 knock-in mice showed deposits between the plasma and basement membrane of the RPE, which altered RPE cell ultra structure. Neurosensory retinal changes included outer segment shortening and detachment from the RPE. Older mice exhibited focal thickening of Bruch's membrane, choriocapillaris degeneration, and outer nuclear layer thinning.3, 12 Abnormal photoreceptor structure was evident in areas with diffuse or focal sub-RPE deposits in our patients. Increasing accumulation in the sub-RPE space might lead to altered RPE function and morphology, which might activate the complement system.3 Impaired RPE function will compromise photoreceptor survival and therefore visual function. Visualising of distinct retinal features using high-resolution imaging provides important information for better understanding of the disease process.
References
Stone EM, Lotery AJ, Munier FL, Heon E, Piguet B, Guymer RH et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet 1999; 22 (2): 199–202.
Michaelides M, Jenkins SA, Brantley Jr MA, Andrews RM, Waseem N, Luong V et al. Maculopathy due to the R345W substitution in fibulin-3: distinct clinical features, disease variability, and extent of retinal dysfunction. Invest Ophthalmol Vis Sci 2006; 47 (7): 3085–3097.
Fu L, Garland D, Yang Z, Shukla D, Rajendran A, Pearson E et al. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum Mol Genet 2007; 16 (20): 2411–2422.
Gaillard MC, Wolfensberger TJ, Uffer S, Mantel I, Pournaras JA, Schorderet DF et al. [Optical coherence tomography in Malattia Leventinese]. Klin Monatsbl Augenheilkd 2005; 222 (3): 180–185.
Souied EH, Leveziel N, Letien V, Darmon J, Coscas G, Soubrane G . Optical coherent tomography features of malattia leventinese. Am J Ophthalmol 2006; 141 (2): 404–407.
Marmor MF, Holder GE, Seeliger MW, Yamamoto S . Standard for clinical electroretinography (2004 update). Doc Ophthalmol 2004; 108 (2): 107–114.
Wojtkowski M, Leitgeb R, Kowalczyk A, Bajraszewski T, Fercher AF . In vivo human retinal imaging by Fourier domain optical coherence tomography. J Biomed Opt 2002; 7 (3): 457–463.
Zawadzki RJ, Jones SM, Olivier SS, Zhao M, Bower BA, Izatt JA et al. Adaptive-optics optical coherence tomography for high-resolution and high-speed 3D retinal in-vivo imaging. Opt Express 2005; 13 (21): 8532–8546.
Zawadzki RJ, Fuller AR, Wiley DF, Hamann B, Choi SS, Werner JS . Adaptation of a support vector machine algorithm for segmentation and visualization of retinal structures in volumetric optical coherence tomography data sets. J Biomed Opt 2007; 12 (4): 041206.
Collins ET . A pathological report upon a case of Doyne's chorioditis (‘Honeycomb’ or ‘family’ choroiditis). The ophthalmoscope 1913; 11: 537–538.
Dusek J, Streicher T, Schmidt K . Hereditary drusen of Bruch's membrane. II: studies of semi-thin sections and electron microscopy results. Klin Monatsbl Augenheilkd 1982; 181 (2): 79–83.
Marmorstein LY, McLaughlin PJ, Peachey NS, Sasaki T, Marmorstein AD . Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum Mol Genet 2007; 16 (20): 2423–2432.
Acknowledgements
The study was supported by National Eye Institute grant 014743 (JSW), the Mira Godard Fund (EH) and the Albrecht Fund (JSW) in collaboration with Bioptigen Inc. We thank Yesmino Elia for study coordination, Cynthia VandenHoven and Carmelina Trimboli for fundus photography and Tom Wright for help with data analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gerth, C., Zawadzki, R., Werner, J. et al. Retinal microstructure in patients with EFEMP1 retinal dystrophy evaluated by Fourier domain OCT. Eye 23, 480–483 (2009). https://doi.org/10.1038/eye.2008.251
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2008.251
Keywords
This article is cited by
-
Doyne honeycomb retinal dystrophy/malattia leventinese induced by EFEMP1 mutation in a Chinese family
BMC Ophthalmology (2018)
-
Gene Therapy Restores Mfrp and Corrects Axial Eye Length
Scientific Reports (2017)
-
Makulopathie mit subretinalen, gelblichen Ablagerungen
Der Ophthalmologe (2011)
