
Abstract
Rare monogenic hyperchylomicronemia is caused by loss-of-function mutations in genes involved in the catabolism of triglyceride-rich lipoproteins, including the lipoprotein lipase gene, LPL. Clinical hallmarks of this condition are eruptive xanthomas, recurrent pancreatitis and abdominal pain. Patients with LPL deficiency and severe or recurrent pancreatitis are eligible for the first gene therapy treatment approved by the European Union. Therefore the precise molecular diagnosis of familial hyperchylomicronemia may affect treatment decisions. We present a 57-year-old male patient with excessive hypertriglyceridemia despite intensive lipid-lowering therapy. Abdominal sonography showed signs of chronic pancreatitis. Direct DNA sequencing and cloning revealed two novel missense variants, c.1302A>T and c.1306G>A, in exon 8 of the LPL gene coexisting on the same allele. The variants result in the amino-acid exchanges p.(Lys434Asn) and p.(Gly436Arg). They are located in the carboxy-terminal domain of lipoprotein lipase that interacts with the glycosylphosphatidylinositol-anchored HDL-binding protein (GPIHBP1) and are likely of functional relevance. No further relevant mutations were found by direct sequencing of the genes for APOA5, APOC2, LMF1 and GPIHBP1. We conclude that heterozygosity for damaging mutations of LPL may be sufficient to produce severe hypertriglyceridemia and that chylomicronemia may be transmitted in a dominant manner, at least in some families.
Similar content being viewed by others


Introduction
Hypertriglyceridaemia (HTG) is a complex phenotype which is may be an independent risk factor for the development of cardiovascular disease.1 In addition to lifestyle factors such as nutrition and physical activity, triglyceride (TG) concentrations are co-determined by genetic factors. Familial lipoprotein lipase deficiency (FLD, MIM 238600) is a rare disease caused by mutations in the LPL gene and is usually considered to follow an autosomal-recessive pattern of inheritance.2 Mutations causative for HTG have been also found in the following genes: APOA5, APOC2, LMF1, GPIHBP1 and GPD1. Apart from this familial disease, recent genome wide association studies (GWAS) and re-sequencing projects have highlighted the contribution of both common and rare sequence variants to the HTG phenotype3, 4, 5, 6, 7 (and reviewed in Kuivenhoven and Hegele8). Wang et al,6 for example, resequenced three candidate genes, LPL, APOC2 and APOA5, in 110 nondiabetic patients with severe HTG (fasting plasma TG>10 mmol/l). This revealed heterozygous mutations in about 10% of the patients, with the LPL gene being most frequently affected from rare variants with functional relevance.6
Among the clinical features of severe hypertriglyceridemia are eruptive xanthomas, hepatosplenomegaly, abdominal pain, episodes of pancreatitis and lipemia retinalis.2 Patients with TG>1000 and >2000 mg/dl have a 5% and 10% to 20% life-time risk for acute pancreatitis, respectively.9 Because of the high risk of pancreatitis due to chylomicronemia, early diagnosis is essential to initiate consequent treatment including simple carbohydrate- and fat-restricted diet, use of medium-chain triglyceride-rich foods and alcohol abstinence. Also therapy with fibrates or nicotinic acid should be provided, even though some rare mutations were associated with a reduced response to oral fibrate therapy6 and in some cases with complete loss of LPL function, fibrate efficacy might lack completely.9 In October 2012, the European Medicine Agency granted the marketing authorization of alipogene tiparvovec for patients with familial lipoprotein lipase deficiency. This medication is a gene therapy for the treatment of FLD patients who have had multiple attacks of acute pancreatitis, despite maximal lipid-lowering medication.10 Evidently, replacement of lipoprotein lipase by gene therapy would be indicated only if variants are present which substantially lower LPL activity. Thus, careful genetic testing is required to identify those who could benefit most from this new therapeutic option.
Materials and methods
Mutation screening
DNA sequence analysis was done by amplification of the coding region of the genes LPL, APOC2, APOA5, LMF1 and GBIHBP1 from genomic DNA followed by direct sequencing of the PCR products. In order to separate the two alleles in the area of the detected sequence variants we generated a PCR product from genomic DNA using oligonucleotides flanking the variant sites: LPL_exon8.F: 5′-AGCTATTTATATTTGGAGAG-3′ and LPL_exon8.R: 5′- CATACAGTAATAAATACAAGGTT-3′. The resulting PCR products were ligated into the vector pCR2.1 and transformed into chemically competent E. coli bacteria using the TOPO TA Cloning Kit (Life Technologies, Rockville, MD, USA). Plasmids from the resulting bacteria clones were extracted using the Plasmid Mini Kit from Qiagen (Hilden, Germany) and the plasmid inserts were sequenced. LPL variant description and exon numbering are based on the NCBI reference sequences NM_000237.2 and NC_000008.11, respectively. The variants identified in the LPL gene were submitted to dbSNP (URL: http://www.ncbi.nlm.nih.gov/SNP/), c.1302A>T, rs587777908 and 1306G>A rs587777909.
Post-heparin LPL activity and mass
EDTA blood (>8 h fasting) was collected 10 min following a heparin bolus (60 units/kg body weight). Plasma samples (patient and healthy control) were used to determine LPL activity using a fluorometric Lipoprotein Lipase (LPL) Activity Assay Kit (Cell Biolabs, Inc., San Diego, CA, USA) according to the manufacturer’s instructions. LPL mass in the plasma samples were determined by an enzyme linked immune sorbent assay (ELISA) using photometric detection (Unilabs, Copenhagen, Denmark). The normal level of LPL mass has been established based on the analysis of a post-heparinized plasma pool from 12 healthy Caucasians. The analytical result is given as the ratio of the actual sample result divided by the normal level (in %).
Results
Clinical case
We report a case of a 57-year-old Caucasian man who was presented to our outpatient lipid clinic at age 54 years with excessive hypertriglyceridemia (TG 4516 mg/dl, 51 mmol/l). His body mass index was 24 kg/m2. Although he had no peripheral stigmata like eruptive xanthoma, he suffered from spleen rupture and had a splenectomy 4 years ago. Secondary causes of severe HTG were ruled out. An impaired glucose tolerance was indicated by an HbA1c of 5.9%. We initiated therapy with micronized fenofibrate (160 mg/day) and omega-3 fatty acids (4000 mg/day). Even after additional lifestyle counseling, alcohol abstinence, restriction of dietary fat, simple carbohydrates and increase of physical activity he still showed markedly elevated triglycerides between 1200 mg/dl (13.6 mmol/l) and 1566 mg/dl (17.7 mmol/l).
Cardiovascular risk factors of our patient include: 40 pack years of nicotine abuse, low HDL-C and positive family history for CVD (the father died from a stroke at the age of 48 years). A sonography of the upper abdomen (at the age of 56 years) showed steatosis of the liver and revealed discrete signs of chronic pancreatitis (hypoechoic and inhomogenous pancreatic parenchyma). Doppler ultrasound showed that he had early atherosclerotic changes with plaques in the right internal carotid artery, as well as in the right and the left common femoral artery. MRI showed discrete microvascular changes of the cerebrum.
While we clearly detected LPL activity in the healthy control sample, we were not able to detect LPL activity in the sample of the patient, both analyzed in post-heparin plasma. Interestingly LPL mass was 140% in the patient sample, when compared with the mass determined from 12 healthy controls (mean).
Molecular genetic analysis
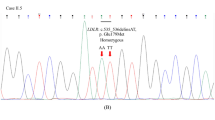
Direct DNA sequencing of the coding sequences of LPL, APOC2, APOA5, LMF1 and GBIHBP1 revealed two new sequence variants in exon 8 of the LPL gene (Figure 1a), whereas no sequence alterations were detected in the other investigated genes. The sequence variants, c.1302A>T and c.1306G>A, are both of the missense type and result in the amino-acid exchanges p.(Lys434Asn) and p.(Gly436Arg), respectively. To the best of our knowledge, neither of the sequence alterations were previously described in humans. Both mutations are located in the carboxy-terminal domain of the LPL protein, which is involved in binding to GPIHBP1, a capillary endothelial cell protein that provides a platform for LPL-mediated processing of chylomicrons.11 Evaluation of the new sequence alterations using ‘PolyPhen-2’ indicated that both variants are ‘probably damaging’, with scores of 0.966 (p.(Lys434Asn)) and 1.0 (p.(Gly436Arg)).12 Similarly, another analysis tool, ‘Mutation Taster’, rated the two sequence alterations as ‘disease causing’,13 suggesting compound heterozygosity for two LPL gene variants. We next ligated an LPL exon 8-specific PCR product generated from the patients DNA into the plasmid pCR2.1, transformed bacteria with the ligation product and sequenced the plasmid inserts from a total of 21 independent clones. Eleven of the plasmid inserts contained the wild-type LPL exon 8 sequence (Figure 1b), whereas the remaining ten plasmid inserts carried the exon 8 sequence with the two sequence alterations, c.1302A>T and c.1306G>A, side by side (Figure 1c). This indicated that the patient has two coexisting variations on the same LPL allele, the other allele is wild type.

Sequence analysis of the LPL variants. (a–c) Segments of genomic DNA sequence from LPL exon 8 of the patient showing (a) the two heterozygous mutations, c.1302A>T and c.1306G>A, detected in the PCR product, (b) the wild-type sequence detected in about the half of the sequenced cloned PCR products and (c) the sequence carrying the two mutations as detected in the other half of the cloned PCR products. (d) Alignment of the LPL amino-acid sequences (single-letter code) from humans and domestic cats in the domain containing the two missense variations detected in the patient. The glycine (G) residue at position 436 and at position 439 mutated in our patient and the cat colony with LPL deficiency is highlighted in red (The alignment was based on UniProt sequences with accession numbers P0685 (human LPL) and P55031 (cat LPL).
Discussion
We report a patient with severe HTG and signs of chronic pancreatitis, steatosis of the liver and premature atherosclerotic changes, but without cutaneous symptoms of HTG. Mutation analysis by direct sequencing and allele separation revealed that the patient carries two sequence alterations within the coding region of the LPL gene coexisting on the same allele, whereas no variation was found in the other LPL gene copy. On the basis of in silico analysis (PolyPhen-2 and Mutation Taster) the two missense variations severely interfere with LPL function.
Interestingly, in members of a domestic cat colony with features of LPL deficiency, homozygous mutations in the LPL gene resulting in the substitution of arginine for glycine at residue 412, the position homologs to the mutated position in our patient, p.(Gly436Arg) (Figure 1d), were identified as the cause underlying the disease.14 Gin et al15 experimentally introduced a mutation into the human LPL open reading frame inserted into a plasmid allowing the expression of the LPL protein with p.(Gly436Arg), the mutation identified in our patient. When ectopically expressed in CHO-K1 cells, the mutant LPL showed increased cleavage at a known furin cleavage site, when compared with the wild-type LPL. The authors showed that the carboxy-terminal part of wild-type LPL was sufficient to bind GPIHBP1, whereas the mutant one (p.(Gly436Arg)) was not. These experimental findings are in line with our in silico analysis and demonstrate that the variation p.(Gly436Arg) identified in our patient is likely of functional relevance. Although the homologous mutation in the LPL-deficient cat results in marginal but significant increase of TGs in heterozygous males, but not in female animals, the HTG in our patient is rather severe. This suggests that the other variant (p.(Lys434Asn)) exerts a strong additive effect on the clinical phenotype. At least to our knowledge, this is the first description of a heterozygous carrier of a LPL variant with TGs in a range otherwise typical for homozygous (or compound heterozygous) LPL deficiency. This might theoretically be due to additive effects of variations in other genes not characterized in the current report, but we consider this possibility not likely as we did not find mutations in APOA5, APOC2, LMF1 and GPIHBP1. Alternatively, FLD may be transmitted in dominant rather than a recessive manner in some families. It is in line with this concept that heterozygous carriers of other LPL missense mutations tend to have elevated triglycerides.16 Interference of the LPL variations identified here with LPL homodimerization might be considered, especially in the light of undetectable LPL activity despite elevated LPL mass measured in the patient post-heparin plasma.
Although our patient has received maximum conventional therapy with diet and lipid-lowering medication, he still presents TGs above 1200 mg/dl (13.6 mmol/l), which puts him at risk to suffer from acute pancreatitis. On the basis of our genetic analysis, however, the patient is not a candidate for the new gene therapy with alipogene tiparvovec, because this therapeutic route is open only for patients with proven homozygosity or compound heterozygosity for mutations in the LPL gene.
In conclusion, we have discovered a so far unknown variant of the LPL gene, a single copy of which is likely sufficient to cause severe HTG. The case presented here also highlights the need for detailed genetic analysis before initiating LPL gene therapy in patients with symptomatic HTG.
References
Murad MH, Hazem A, Coto-Yglesias F et al: The association of hypertriglyceridemia with cardiovascular events and pancreatitis: a systematic review and meta-analysis. BMC Endocr Disord 2012; 12: 2.
Hegele RA, Ginsberg HN, Chapman MJ et al: The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diab Endocrinol 2014; 2: 655–666.
Surendran RP, Visser ME, Heemelaar S et al: Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med 2012; 272: 185–196.
Teslovich TM, Musunuru K, Smith AV et al: Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466: 707–713.
Wang J, Ban MR, Zou GY et al: Polygenic determinants of severe hypertriglyceridemia. Hum Mol Genet 2008; 17: 2894–2899.
Wang J, Cao H, Ban MR et al: Resequencing genomic DNA of patients with severe hypertriglyceridemia (MIM 144650). Arterioscler Thromb Vasc Biol 2007; 27: 2450–2455.
Wright WT, Young IS, Nicholls DP, Graham CA : Genetic screening of the LPL gene in hypertriglyceridaemic patients. Atherosclerosis 2008; 199: 187–192.
Kuivenhoven JA, Hegele RA : Mining the genome for lipid genes. Biochimica et Biophysica Acta 2014; 1842: 1993–2009.
Scherer J, Singh VP, Pitchumoni CS, Yadav D : Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol 2014; 48: 195–203.
Gaudet D, Methot J, Kastelein J : Gene therapy for lipoprotein lipase deficiency. Curr Opin Lipidol 2012; 23: 310–320.
Beigneux AP, Davies BS, Gin P et al: Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab 2007; 5: 279–291.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D : MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
Ginzinger DG, Lewis ME, Ma Y, Jones BR, Liu G, Jones SD : A mutation in the lipoprotein lipase gene is the molecular basis of chylomicronemia in a colony of domestic cats. J Clin Investig 1996; 97: 1257–1266.
Gin P, Goulbourne CN, Adeyo O et al: Chylomicronemia mutations yield new insights into interactions between lipoprotein lipase and GPIHBP1. Hum Mol Genet 2012; 21: 2961–2972.
Johansen CT, Wang J, Lanktree MB et al: Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet 2010; 42: 684–687.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
UK received honoraria for lectures from Fresenius medical care, Sanofi and B. Braun. The remaining authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kassner, U., Salewsky, B., Wühle-Demuth, M. et al. Severe hypertriglyceridemia in a patient heterozygous for a lipoprotein lipase gene allele with two novel missense variants. Eur J Hum Genet 23, 1259–1261 (2015). https://doi.org/10.1038/ejhg.2014.295
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.295
