
Abstract
The imprinted domain in human 15q11-q13 is controlled by a bipartite imprinting centre (IC), which overlaps the 5′ part of the paternally expressed SNURF–SNRPN gene. We have recently described two novel genes upstream of SNURF–SNRPN (PWRN1 and PWRN2), which are biallelically expressed in the testis. We have now found that PWRN1 represents an alternative 5′ part of SNURF–SNRPN, and that its expression in the brain is imprinted. To determine when the locus is activated during spermatogenesis and which factors are involved in this process, we have mined gene-expression data of testicular biopsies from men with different types of spermatogenic failure. Whereas PWRN1–SNURF–SNRPN and PWRN2 are expressed in post-meiotic germ cells only, a hitherto undetected SNURF–SNRPN upstream transcript is expressed already at meiosis. Several epigenetic factors (eg, MBD1 and MBD2 isoforms, MBD3L1, SUVH39H2, BRDT, and EZH2) are upregulated at specific stages of spermatogenesis, suggesting that they play an important role in the epigenetic reprogramming during spermatogenesis.
Similar content being viewed by others


Introduction
Human chromosome 15q11-q13 harbors a 2-Mb imprinted domain, which is regulated by a bipartite imprinting centre (for a review see Horsthemke and Wagstaff1). One element of the imprinting centre (IC) is defined by the shortest region of deletion overlap in patients with Prader–Willi syndrome and a paternal imprinting defect (PWS-SRO). The other element is defined by the shortest region of deletion overlap in patients with Angelman syndrome and a maternal imprinting defect (AS-SRO). The two elements are ∼30 kb apart (see Figure 1a).

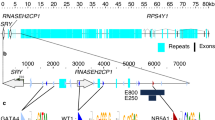
Physical map. (a) Schematic overview of the ∼800 kb chromosomal region from PWRN2 to SNURF–SNRPN. Not drawn to scale. The start and orientation of transcription are indicated by arrows. AS-SRO and PWS-SRO, the two elements of the bipartite imprinting centre. Exons are indicated as black boxes. U6, U7, and U8 are unpublished SNURF–SNRPN upstream exons (U6, Chr15: 22.717.397–22.717.495; U7, Chr.15: 22.725.258–22.725.314; U8, Chr.15: 22.746.815–22.746.930). (b) Published EST clones and mRNA fragments. (c) RT-PCR clones generated in this study.
The IC overlaps the 5′ part of the paternally expressed SNURF–SNRPN gene, which has at least three alternative start sites and many alternative exons.2, 3 In 2000 we identified C15orf2, which is a 7.5 kb intronless gene located ∼300 kb upstream of the major SNURF–SNRPN start site.4 C15orf2 has a 3.5 kb open reading frame encoding an 1156 amino-acid protein of unknown function. Recently we have identified two novel genes (PWRN1 and PWRN2), which are located centromeric to C15orf25 and which do not appear to have any protein-coding potential. PWRN1, PWRN2 and C15orf2 are mainly expressed in the testis, where expression is biallelic. PWRN1 and C15orf2 are also expressed in the brain. The finding of monoallelic expression in the brain suggested that these genes are subject to genomic imprinting.5 Here we show that PWRN1 is part of the SNURF–SNRPN transcription unit and that expression in the brain is from the paternal allele only.
The mechanism by which the IC controls imprinting in 15q11-q13 is unknown. The overlap of the IC with the 5′ part of the SNURF–SNRPN locus suggests that the transcripts may play a role in this process, although there is no experimental evidence for this to date. Based on the phenotypic consequences of microdeletions affecting the PWS-SRO element of the IC, the AS-SRO element of the IC, or both, we have proposed that the maternal pattern of epigenetic modification, with CpG methylation of MKRN3, NDN and SNURF–SNRPN promoters and silencing of those genes, is the default state of the imprinted domain, that the PWS-SRO is unconditionally required for a chromosome to have the paternal pattern of epigenetic modification and gene expression, and that the AS-SRO is required for a chromosome to have the maternal pattern, if the chromosome has an intact PWS-SRO.1 In other words, the PWS-SRO acts to activate the paternal copy of the imprinted domain or keep this copy active,6 and the AS-SRO counteracts this activity.
As the AS-SRO contains two SNURF–SNRPN upstream exons, we and others have proposed that these transcripts may play a role in methylating the PWS-SRO and silencing the region in the female germline.2, 7 This hypothesis is supported by the work of Chotalia et al.,8 who found that transcription is required for the establishment of germline-methylation marks at imprinted genes. On the other hand, transgene experiments in which the 880 bp AS-SRO was found to be sufficient for silencing the SNURF–SNRPN promoter seem to argue against this hypothesis.9 Alternatively, the upstream transcripts may play a role in activating the locus, and the AS-SRO may contain a silencer, which represses transcription in the maternal germline. In any case, it is clear that trans-acting factors must be involved.
To lay the foundation for answering these questions, we have investigated the expression patterns of the SNURF–SNRPN upstream transcripts, as well as of 20 genes encoding epigenetic factors. Owing to the limited availability of normal human testis samples and technical problems associated with RNA in situ hybridization of this material, we have mined gene-expression data of testicular biopsies from men with different types of spermatogenic failure (complete tubular atrophy, Sertoli-cell only syndrome, arrest before meiosis, arrest at meiosis, arrest at round spermatids, and uniform hypospermatogenesis) and with full spermatogenesis.10 We show that these data can be used to infer gene-expression levels in spermatogonia, spermatocytes and round spermatids.
Materials and methods
Preparation of DNA and RNA
Total RNA and DNA from the fetal brain were extracted using the RNeasy Mini Kit and the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's recommendations.
Exon connection PCR and RT-PCR
Exon connection PCR was performed on human cDNA from the testis and the fetal brain (Marathon-Ready cDNA, Clontech, Heidelberg, Germany) with the following primers: SNRPN_Exon23 (5′-TCCTAACAACCACTATAAGCTTGG-3′)+SNRPN_4_rw (5′-GATGCTTCACGGTTTTCACA-3′) (annealing temperature 58°C) and SNRPN_4_fw (5′-CCCACAAGTAGGGTTGACCA-3′)+SNRPN_U5 (5′-CAGATTCTTCTGCCGCAAAC-3′) (annealing temperature 58°C).
RT-PCR for characterization of mRNA BC035402 was performed on human testis cDNA (Marathon-Ready cDNA, Clontech) with the following primers: BC035402_fw1 (5′-TTAACCTGCGTTTTCCCGTA-3′)+BC04ex_rw (5′-AAACACAACACCCCAAGCAT-3′) (annealing temperature 60°C) and BC035402_fw1 (5′-TTAACCTGCGTTTTCCCGTA-3′)+BC04exnest_rw (5′-TGTTGCAAGGAACGAGACTG-3′) (annealing temperature 60°C).
PCR products were re-amplified, subcloned into the pGEM-T Easy vector (Promega, Mannheim, Germany) and sequenced with specific primers. In the case of multiple PCR products, the products were excised from the gel, purified with the MinElute Gel Extraction Kit (Qiagen) and sequenced directly or sequenced with specific primers after subcloning into the pGEM-T Easy vector.
Sequence analysis
Sequencing reactions were carried out with fluorescence-tagged dideoxynucleotides (BIG Dye kit v1.1, Applied Biosystems, Foster City, CA, USA) and the Taq cycle sequencing procedure. Sequences were analysed on an ABI 3100 DNA sequencer.
Allele-specific expression analysis
DNA from the fetal brain (cerebrum) and corresponding parental DNA samples from blood were analysed for an expressed SNP (rs12908526) in exon 26 of PWRN1 by PCR. For sequencing, the following primers were used: Ex26SNPF (5′-GGAACAAGTTTATTCACACACAGA-3′) and Ex26SNPR (5′-TGTCAACAGGGAAAGTGGAA-3′) (annealing temperature 58°C). RNA from the same fetal brain was reverse transcribed to cDNA with random hexamers and also analysed for SNP rs12908526. To check the integrity of the RNA and to exclude contamination with the genomic DNA, we used primers for the ß-actin gene as a control.5
Expression profiles
The expression data of spermatogenesis genes, genes encoding DNA and histone-binding and -modifying proteins, as well as the SNURF–SNRPN upstream transcripts were extracted from the dataset published by Spiess et al.10 To corroborate the findings, which were obtained with Affymetrix GeneChip microarrays (Affymetrix, Santa Clara, CA, USA), we included expression data acquired with a completely different platform based on single-oligonucleotide probes (Codelink system, Applied Microarray Inc., Tempe, AZ, USA) in the analysis.11 This was done by building an intersection of the two platforms using HUGO gene abbreviations. Hierarchical cluster analysis was carried out with the mean expression values from the different stages of spermatogenic impairment and clustered with the TIGR MeV software v4.3 (The TIGR Consortium, http://www.tm4.org/mev.html) using Manhattan distance and average linkage. Genes that were common to both platforms (GeneChip and Codelink), and which displayed similar expression patterns, were denoted with one asterisk on the heatmap and two asterisks if expression patterns were not concordant.
Results
PWRN1 is part of the IC–SNURF–SNRPN transcription unit
A database search (UCSC browser hg18, http://genome.ucsc.edu/cgi-bin/hgGateway) for expressed sequences upstream of SNURF–SNRPN revealed the presence of two overlapping spliced EST clones, DB525036 and BI825340. DB525036 consists of five exons and BI825340 consists of four exons. BI825340 shares two exons with DB525036, but contains two additional exons not present in EST DB525036 (see Figure 1b). By detailed sequence comparison we found that the most upstream exon of EST DB525036 represents exon 23 of PWRN1. PWRN1 has no protein-coding potential and is subject to alternative splicing and polyadenylation.5 As DB525036 lacks exons 24–26 of PWRN1, but has additional four exons downstream to exon 26, it represents a novel splice variant, which extends more telomeric. Interestingly, the novel exons flank C15orf2, which lies now within an intron of PWRN1. By exon connection PCR we were able to verify the novel exons in fetal brain cDNA and could also identify one additional exon (see RT1 in Figure 1c). The most telomeric novel exon maps only 285 bp proximal to U1B, which represents one of the two known alternative start sites of the SNURF–SNRPN upstream transcripts. PWRN1 transcripts display the same complex splicing pattern as previously observed for the upstream transcripts of SNURF–SNRPN, thus it was possible that the PWRN1 exons represent an additional part of the complex IC–SNURF–SNRPN transcription transcripts. To find out whether this is true we performed exon connection PCR on cDNA of the fetal brain and the testis with primers that anneal in SNURF–SNRPN exons U2 and U5 and in the last but one novel downstream exon of PWRN1. Indeed, we could connect PWRN1 with the upstream exon U5 in both tissues (see RT3 and RT4 in Figure 1c). Furthermore, we found in testis cDNA a novel exon between U3 and U4 (see RT4 in Figure 1c).
PWRN1 expression is imprinted in the brain
We have recently reported that PWRN1 shows monoallelic expression in the fetal brain. However, as parental DNA samples were not available at that time, we could not distinguish whether expression is restricted to the paternal or maternal allele. In the meantime, we were able to obtain DNA and RNA samples from fetal brain and parental DNA samples. Analysis of an expressed single-nucleotide repeat (SNP, rs12908526, A/T) in exon 26 of PWRN1 showed heterozygosity (A/T) in fetal brain DNA (Figure 2). The mother was homozygous for the A allele and the father was homozygous for the T allele. In the fetal brain RNA only the T allele was detected, indicating that monoallelic expression is paternal. These findings suggest that expression is imprinted.

Paternal expression of PWRN1. Sequence analysis of DNA samples from mother, father, and fetus (upper three panels) around a single-nucleotide polymorphism in exon 26 (rs12908526). The fetus is heterozygous, but expresses only the paternal (T) allele (lower panel).
Expression profiles
The finding of biallelic expression of the SNURF–SNRPN upstream transcripts in the testis raised three questions: (i) Are these transcripts expressed in somatic cells or in germ cells? (ii) If they are expressed in germ cells, at which stage of spermatogenesis are they expressed? (iii) Which epigenetic factors regulate the expression? To address these questions we have mined gene-expression data of testicular biopsies from men with different types of spermatogenic failure as determined with Affymetrix GeneChips.10 The arrest types used in this study differ essentially in their germ-cell composition so that data can be extracted by filtering genes that pertain to those germ-cell types that appear from a higher impaired pathology to a lower one. In detail, this would be the following (using the abbreviations defined in the legend to Figure 3)

Heatmap of the mean expression values of selected genes in each group of men as determined on the Affymetrix GeneChip platform. Types of spermatogenic failure (number of individuals): CTA, complete tubular atrophy (n=1); SCO, Sertoli-cell only (n=7); PMA, premeiotic arrest of spermatogenesis (n=3); MA, meiotic arrest of spermatogenesis (n=5); RS, presence of round spermatids (n=4); HYS, hypospermatogenesis (n=7); FS, full spermatogenesis (n=8). Dark blue indicates no expression (log 2=2), yellow a high expression (log 2=10). (a) Expression of four spermatogenesis genes. (b) Hierarchical cluster analysis reveals four clusters (1–4). Note that several genes are represented by different probe sets. The expression pattern of genes marked with one asterisk could be confirmed by a different microarray platform based on single-oligonucleotide probes (Codelink, see Materials and methods). The two asterisks denote different patterns between the two platforms.
CTA => SCO: appearance of Sertoli cells
SCO => PMA: appearance of spermatogonia (pre-meiotic arrest)
PMA => MA: appearance of primary/secondary spermatocytes (meiotic arrest)
MA => RS: appearance of round spermatids
RS => HYS: appearance of low amounts of elongated spermatids and testicular spermatozoa
To determine whether these data can be used to determine the upregulation of genes at specific stages of spermatogenesis, we examined several spermatogenesis genes. The synaptonemal complex protein 3 (SYCP3) is involved in the pairing of homologous chromosomes during meiosis I. As shown in Figure 3a, the SYCP3 gene is expressed at very low levels in the testis lacking germ cells (CTA and SCO). Low expression was observed in men with spermatogenic arrest before meiosis (PMA), and high expression was observed in men with spermatogenic arrest at meiosis (MA) and at the round spermatid stage (RS), as well as in hypo- (HYS) and normospermic (FS) men. Transition protein 1 (TNP1) as well as the protamines PRM1 and PRM2 are involved in the packaging of DNA after meiosis into the sperm nucleus. As expected, these genes are expressed at high levels only in men with spermatogenic arrest at the round spermatid stage, as well as in hypo- and normospermic men.
These data show that the expression profiles of testicular biopsies from men with spermatogenic failure can be used to determine the stage-specific upregulation of genes during spermatogenesis. Therefore, we used this dataset to analyze the developmental expression profile of the SNURF–SNRPN upstream transcripts and genes encoding DNA and histone-binding and -modifying proteins. Several genes are represented by two or more probe sets on the microarray, and in some cases the different probe sets detect different spliceforms (isoforms).
Using hierarchical cluster analysis (Figure 3b), we identified four clusters of genes. Cluster 1 contains genes with little difference in expression (DNMT2, DNMT3A, DNMT3B, DNMT3L, MBD3, MBD3L2, MBD4, MBD5, MBD6, MECP2, SUV39H1, CTCF, and isoforms of MBD1 and MBD2). Some of these genes tend to be expressed at a lower level in men with late spermatogenic arrest or normal spermatogenesis, indicating that the expression is mainly in somatic cells. We did not find any stage-specific differences in the expression of histone deacetylases (HDCAs) either (not shown). Cluster 2 contains only one gene (DNMT3L), which is below the level of detection.
Cluster 3 contains genes that are mainly expressed in germ cells and upregulated at late meiosis or after meiosis. This cluster includes the spermatogenesis genes TNP1, PRM1 and PRM2, as well as MBD1, MBD3L1, BRDT, and several IC–SNURF–SNRPN transcripts.
The remaining genes form a somewhat heterogeneous cluster (Cluster 4). Most of these genes are upregulated before or at early meiosis. This cluster includes the spermatogenesis gene SYCP3, as well as DNMT1, MBD1, MBD2, MBD4, MDB6, SUV39H2, EZH2, CTCFL, and one IC–SNURF–SNRPN transcript. Among the epigenetic genes of this cluster, MBD2, EZH2, SUV39H2 and CTCFL transcripts are clearly germ cell-specific and upregulated before meiosis. A detailed analysis of the histone methyltransferase genes SUV39H1, SUV39H2 and EZH2 is shown in Figure 4a.

Detailed diagrams of gene-expression values in individual biopsies. Each position on the x-axis represents one individual. The background of the expression is between log 2=3–4. Abbreviations are as in Figure 3. (a) Expression profile of SUV39H1 (green), SUV39H2 (magenta), and EZH2 (blue). (b) Expression of the MBD1 isoforms (a, magenta; b, blue). Below the diagram is an overview of the different splice forms as given in the UCSC browser hg18. The two probe sets detect alternative 3′ exons. (c) Expression of the short (blue) and the long isoform (magenta) of MBD2. The location of the probe sets is indicated below the diagram. (d) Expression of PWRN2 (blue), PWRN1 (magenta), and the mRNA BC035402 (green), which maps between the SNURF–SNRPN upstream exons U1B* and U1A. The location of the probe sets is indicated below the diagram.
To corroborate these findings, genes displayed in the heatmap of Figure 3 were interrogated on a dataset from a completely different, single-oligonucleotide microarray platform11 by using the intersection obtained from HUGO annotations. This cross-platform comparison validates the expression changes described here and obviates the need to validate single genes, as has been shown in a study in which 93% of genes common to two microarray platforms could be confirmed in their differential expression by quantitative real-time PCR12. Similar to the results described there, we also observed a high concordance in the gene-expression profiles obtained from the two platforms (16 out of 18; see asterisks in Figure 3).
MBD1, MBD2 and IC-SNURF-SNRPN are represented by different probe sets, which revealed different patterns of expression. The detailed expression pattern and the location of the MBD1 and MBD2 probe sets are shown in Figure 4b and Figure 4c, respectively. This analysis shows that the testis-specific isoforms of MBD1 and MBD2 use alternative 3′ exons. The premeiotic form of MBD2 is a truncated form.
As shown in Figure 4d, PWRN1 and PWRN2 are upregulated after meiosis. In contrast, the transcript containing the mRNA BC035402 is upregulated at meiosis. This mRNA represents no known upstream exon of SNURF–SNRPN, and we failed to link it to any of the known upstream exons in the testis, the fetal brain and leukocyte cDNA. Although the mRNA is colinear with genomic DNA, we identified two spliced alternative isoforms by RT-PCR of testis cDNA (data not shown).
Discussion
As shown by Spiess et al.,10 the microarray-based analysis of mRNA levels in testicular biopsies from fertile men and from men with different types of spermatogenic failure is highly reliable: in 19 of 20 genes tested, the expression patterns could be confirmed by quantitative RT-PCR.10 For validation of the gene-expression data described here, we used a second, independent dataset of human spermatogenesis.11 Except for EZH2 and MBD1, concordant expression profiles were found. The apparent discrepancy regarding the latter two genes may, at least in the case of MBD1, be because of different probes on the two array platforms and the presence of different splice forms (see below). The validity of our data is further supported by the finding that the expression values of different individuals within each disease group are very similar, with the exception of hypospermatogenesis (see Figure 4). The latter group is likely to be etiologically heterogeneous, and the expression patterns may help to classify these patients further. Last, but not least, in those cases in which mouse data are available, the gene-expression profiles described here are in complete concordance with the gene-expression profiles observed in the mouse (see below).
Here we have used the data to investigate the stage-specific upregulation of the SNURF–SNRPN locus as well as epigenetic regulator genes during male germ-cell development. The expression patterns of the spermatogenesis genes SYCP3, TNP1, PRM1 and PRM2 showed that this is a valid approach. It should be noted, however, that this approach has two limitations. (i) It is not possible to analyze genes expressed in germ cells and somatic cells, unless there is a germ-cell-specific isoform, because the presence of somatic transcripts in the biopsies masks possible differences in the abundance of germ-cell-specific transcripts. (ii) It is not possible to study the stage-specific downregulation of genes, because each biopsy contains cells at different stages of the germ-cell development. For example, biopsies from men with arrest at the round spermatid stage contain all stages of spermatogonia and spermatocytes. Thus, a gene that is active in spermatocytes but silenced in round spermatids will show up in these biopsies, as well as in biopsies from men with hypospermatogenesis and full spermatogenesis.
We have recently reported the identification of two novel genes upstream of SNURF—SNRPN.5 Here we show that PWNR1 is not an independent gene, but an alternative 5′ part of the SNURF–SNRPN gene. Similar to other transcripts made from this locus, expression in brain is imprinted and from the paternal chromosome only. It was unclear why PWNR1 and PWRN2, which is transcribed from the opposite strand, are biallelically expressed in the testis. Here we show that both transcripts are made in post-meiotic germ cells, that is, from the haploid genome after the establishment of the paternal imprint. If they play a role in imprinting, they may not have a role in establishing the paternal imprint, but in maintaining the paternal allele in an open chromatin configuration.
In contrast to the PWRN1 and PWRN2 transcripts, the transcript containing the mRNA BC035402 is upregulated at meiosis. We have not been able to link this transcript to other SNURF–SNRPN upstream exons, and thus the identity of this transcript remains elusive.
To determine which epigenetic factors might be involved in upregulating the upstream transcripts and how this relates to the timing of epigenetic reprogramming during spermatogenesis,13 we have analysed the expression patterns of several epigenetic regulator genes. Our results are in agreement with and extend previous findings in the mouse and/or humans. We confirm in humans that CTCFL is a germ-cell-specific factor that is upregulated before meiosis. As shown in the mouse by Loukinov et al.,14 CTCFL is expressed in a mutually exclusive manner with CTCF during male germ-cell development, and the erasure of methylation marks during male germ-line development is associated with dramatic up-regulation of CTCFL and down-regulation of CTCF expression. This makes CTCFL a prime candidate protein for the epigenetic reprogramming factor acting in the male germ line.
SUV39H2, which encodes a testis-specific histone H3 lysine 9 methyltransferase related to SUV39H1, is upregulated during meiosis. Immunolocalization of the murine Suv39h2 protein during spermatogenesis by O′Carroll et al.,15 indicated enriched distribution at the heterochromatin from the leptotene to the round spermatid stage.
BRDT is a testis-specific member of the distinctive BET sub-family of bromodomain motif-containing proteins, a motif that binds acetylated lysines and is implicated in chromatin remodeling. As shown by Shang et al.,16 in the mouse, its expression is restricted to the germ line, specifically to pachytene and diplotene spermatocytes and early spermatids. Our findings are consistent with observations in the mouse.
MBD3L1 is a methyl-cytosine binding transcriptional repressor. As reported earlier by Jiang et al.17 and suggested by our findings, the expression of MBD3L1 is observed only in round spermatids, suggesting a role for the gene product in the postmeiotic stages of male germ-cell development.
We confirm that the MBD2 gene encodes at least two isoforms of a methyl-CpG binding domain protein, one of which is testis-specific.18 It was unknown, however, when the testis-specific isoform is expressed. Here we show that it is expressed very early during spermatogenesis, before the meiosis. The function of MBD2 has been a matter of dispute. Bhattacharya et al.19 have suggested that it acts as a DNA demethylase and is thus a transcriptional activator. However, no other group has been able to reproduce this finding. In fact, Ng et al.20 have found strong evidence that MBD2 acts as transcriptional repressor. Both isoforms contain the methyl-CpG binding domain. It is tempting to speculate that the short, testis-specific isoform lacks repressor activity and demethylates DNA. If so, our findings suggest that it may be involved in erasing methylation patterns in primordial germ cells or spermatogonia.
We show for the first time that the male germ line expresses an MBD1 isoform, which is derived from an alternative 3′ exon. In contrast to the above-mentioned isoform of MBD2, it is not shorter than the somatic isoform and is expressed somewhat later than MBD2. We also show for the first time that EZH2, which encodes a Polycomb group protein involved in methylating histone H3 at lysine 27, is expressed very early during spermatogenesis.
In summary, we have found that several epigenetic factors (MBD1 and MBD2 isoforms, MBD3L1, SUVH39H2, BRDT, and EZH2) are upregulated at specific stages of human spermatogenesis, suggesting that they play an important role in the epigenetic reprogramming of the genome during spermatogenesis. More studies are necessary to elucidate all the cis- and trans-acting factors involved in activating the paternal copy of 15q11-q13.
References
Horsthemke B, Wagstaff J : Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A 2008; 146A: 2041–2052.
Dittrich B, Buiting K, Korn B et al: Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat Genet 1996; 14: 163–170.
Färber C, Dittrich B, Buiting K, Horsthemke B : The chromosome 15 imprinting centre (IC) region has undergone multiple duplication events and contains an upstream exon of SNRPN that is deleted in all Angelman syndrome patients with an IC microdeletion. Hum Mol Genet 1999; 8: 337–343.
Färber C, Gross S, Neesen J, Buiting K, Horsthemke B : Identification of a testis-specific gene (C15orf2) in the Prader-Willi syndrome region on chromosome 15. Genomics 2000; 65: 174–183.
Buiting K, Nazlican H, Galetzka D, Wawrzik M, Gross S, Horsthemke B : C15orf2 and a novel noncoding transcript from the Prader-Willi/Angelman syndrome region show monoallelic expression in fetal brain. Genomics 2007; 89: 588–595.
El-Maarri O, Buiting K, Peery EG et al: Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat Genet 2001; 27: 341–344.
Mapendano CK, Kishino T, Miyazaki K et al: Expression of the Snurf-Snrpn IC transcript in the oocyte and its putative role in the imprinting establishment of the mouse 7C imprinting domain. J Hum Genet 2006; 51: 236–243.
Chotalia M, Smallwood SA, Ruf N et al: Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev 2009; 23: 105–117.
Shemer R, Hershko AY, Perk J et al: The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat Genet 2000; 26: 440–443.
Spiess AN, Feig C, Schulze W et al: Cross-platform gene expression signature of human spermatogenic failure reveals inflammatory-like response. Hum Reprod 2007; 22: 2936–2946.
Feig C, Kirchhoff C, Ivell R, Naether O, Schulze W, Spiess AN : A new paradigm for profiling testicular gene expression during normal and disturbed human spermatogenesis. Mol Hum Reprod 2007; 13: 33–43.
Bosotti R, Locatelli G, Healy S et al: Cross platform microarray analysis for robust identification of differentially expressed genes. BMC Bioinformatics 2007; 8: 1–S5.
Sasaki H, Matsui Y : Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet 2008; 9: 129–140.
Loukinov DI, Pugacheva E, Vatolin S et al: BORIS, a novel male germ-line-specific protein associated with epigenetic reprogramming events, shares the same 11-zinc-finger domain with CTCF, the insulator protein involved in reading imprinting marks in the soma. Proc Natl Acad Sci USA 2002; 99: 6806–6811.
O′Carroll D, Scherthan H, Peters AH et al: Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol 2000; 20: 9423–9433.
Shang E, Salazar G, Crowley TE, Wang X, Lopez RA, Wolgemuth DJ : Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene Expr Patterns 2004; 4: 513–519.
Jiang CL, Jin SG, Lee DH et al: MBD3L1 and MBD3L2, two new proteins homologous to the methyl-CpG-binding proteins MBD2 and MBD3: characterization of MBD3L1 as a testis-specific transcriptional repressor. Genomics 2002; 80: 621–629.
Hendrich B, Bird A : Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol 1998; 18: 6538–6547.
Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M : A mammalian protein with specific demethylase activity for mCpG DNA. Nature 1999; 397: 579–583.
Ng HH, Zhang Y, Hendrich B et al: MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet 1999; 23: 58–61.
Acknowledgements
We thank Professor C Kirchhoff for helpful discussions and the Deutsche Forschungsgemeinschaft for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ethical statement
This study was approved by the local ethic committee.
Rights and permissions
About this article
Cite this article
Wawrzik, M., Spiess, AN., Herrmann, R. et al. Expression of SNURF–SNRPN upstream transcripts and epigenetic regulatory genes during human spermatogenesis. Eur J Hum Genet 17, 1463–1470 (2009). https://doi.org/10.1038/ejhg.2009.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.83
Keywords
This article is cited by
-
Increased copy number of imprinted genes in the chromosomal region 20q11-q13.32 is associated with resistance to antitumor agents in cancer cell lines
Clinical Epigenetics (2022)
-
Construction and analysis of a lncRNA (PWRN2)-mediated ceRNA network reveal its potential roles in oocyte nuclear maturation of patients with PCOS
Reproductive Biology and Endocrinology (2018)
-
Aberrant expression of long noncoding RNAs in cumulus cells isolated from PCOS patients
Journal of Assisted Reproduction and Genetics (2016)
-
Copy Number Variations at the Prader‐Willi Syndrome Region on Chromosome 15 and associations with Obesity in Whites
Obesity (2011)
-
The C15orf2 gene in the Prader–Willi syndrome region is subject to genomic imprinting and positive selection
neurogenetics (2010)
