
Abstract
Anophthalmia (no eye), microphthalmia (small eye) and associated ocular developmental anomalies cause significant visual handicap. In most cases the underlying genetic cause is unknown, but mutations in some genes, such as SOX2, cause ocular developmental defects, particularly anophthalmia, in a subset of patients. Here, we describe a four-generation family with a p.Asp123Gly mutation in the highly conserved partner-factor interaction region of the SOX2 protein, which is important for cell-specific actions of SOX2. The proband in this family has bilateral anophthalmia and several other family members have milder ocular phenotypes, including typical optic fissure coloboma. Expression studies indicate that Sox2 is expressed in the eye at the site of closure of the optic fissure during development. The SOX2 mutation in this family implicates the partner-factor interaction region of SOX2 in contributing to the specificity of SOX2 action in optic fissure closure. Our findings indicate that investigation of SOX2 in a broad range of eye anomaly patients aids in the determination of particular functions of SOX2 in development.
Similar content being viewed by others


Introduction
Anophthalmia and severe microphthalmia with associated developmental ocular conditions including sclerocornea, cataract and ocular coloboma, are conditions with significant morbidity. The underlying disease genes and variability of expression of associated ocular features are poorly understood. Heterozygous SOX2 mutations are present in approximately 10% of patients with anophthalmia and severe microphthalmia.1 Mutations in other genes have been identified in anophthalmia/microphthalmia patients including, RAX, PAX6 and OTX2,2, 3, 4 but these are less frequently found. CHX10 mutations have also been identified, and these are particularly seen in one ethnic group.5 STRA6 mutation patients have syndromic associations including diaphragmatic defects.6 For a patient who may have an ocular condition such as anophthalmia, microphthalmia, coloboma, sclerocornea or cataract, siblings and children may also be affected. For those with a less severe ocular anomaly, there is also the possibility of a more severe ocular phenotype if recurrence occurs.7
The most commonly described ocular phenotype associated with SOX2 mutations is that of a severe bilateral eye defect such as anophthalmia or severe microphthalmia. If there is an eye present, associated ocular features may include sclerocornea and retinal detachment. Colobomata are infrequently reported and when present are usually described as atypical or optic nerve colobomata rather than typical optic fissure closure defects.8, 9 Associated extraocular features include intrauterine growth retardation, poor growth postnatally with pituitary insufficiency, developmental delay, seizures, sensorineural hearing loss and male genital abnormalities. SOX2 mutations have also been identified in some patients with anophthalmia-oesophageal genital syndrome.10, 11 SOX2 mutations most commonly code for stop codons or cause frameshifts, which are predicted to lead to haploinsufficiency and loss of function of the protein.1, 9 Missense mutations are rare8 and none have so far been reported in the partner-factor-binding region, which gives cell-specificity to the actions of SOX2.12 Most SOX2 mutations arise de novo with recurrence usually attributed to gonosomal mosaicism in a parent,10, 13 although non-penetrance has also been reported.14
Materials and methods
Mutation detection
Genomic DNA extracted from whole blood was used for PCR amplification. In patient II.9 (Figure 1a), the coding exon and adjacent flanking regions of SOX2 (GenBank accession number NM_003106) were amplified by PCR using two overlapping primer sets (primer sequences and PCR conditions available on request). Amplified products were purified using Exonuclease I (Epicentre, Madison, WI, USA) and rapid alkaline phosphatase (Roche, Mannheim, Germany) and sequenced bi-directionally using standard techniques (Big Dye TM terminator, Applied Biosystems, Foster City, CA, USA). A novel mutation was identified and segregation of the mutation was investigated by direct sequencing of genomic DNA from other family members. The more severely affected members in the family were also investigated for variation in other genes associated with anophthalmia and microphthalmia, using PCR amplification and direct sequencing of the coding exons and flanking intronic sequences of OTX2 (NM_021728), CHX10 (NM_182894), RAX (NM_013435) and PAX6 (NM_001604). Nucleotide and codon numbering refers to the coding DNA sequence starting at the translation initiation site, with +1 as the A of the ATG start codon. Written informed consent was obtained from patients, in accordance with ethics approval from the Children's Hospital at Westmead Ethics Committee.

Pedigree and clinical features of family. (a) In this four-generation pedigree, individuals with the SOX2 mutation, p.Asp123Gly, are identified by an asterisk, and those without are identified by a minus sign. (b) Left eye of patient I.2 shows prominent posterior embryotoxon. Left eye of patient II.1 shows inferior iris stromal thinning. Left eye of patient II.5 shows optic nerve hypoplasia. Left eye of patient II.9 shows a subtle inferior pupillary defect. Patient III.1 has bilateral chorioretinal and iris colobomata. Patient III.3 has a small retinal tuft inferiorly in the left eye. Patient IV.2 had bilateral clinical anophthalmia and in the image there are bilateral prostheses in place.
Animal tissues and section in situ hybridization
Mouse embryos at embryonic days 10.5 (E10.5) and E11.5 were fixed in 4% paraformaldehyde overnight, placed in 20% sucrose solution and embedded in OCT compound (Tissue Tek, ProSciTech, Kirwan, Australia), frozen on dry ice and stored at −80°C until required. Cryostat sections were cut at 12-μm thickness in a parasagittal plane. Using AmpliScribe Transcription Kits (Astral Scientific, Sydney, Australia) and digoxigenin-11-UTP (Roche), Sox2 sense and antisense riboprobes were synthesized from the plasmid kindly provided by Dr Lovell-Badge, MRC, UK. The in situ hybridization was conducted as described earlier for digoxygenin-labeled riboprobes on cryosections.15 Transcripts were detected using an anti-digoxygenin alkaline phosphatase-linked antibody (Roche) and visualized using BM purple (Roche).
Results
Clinical findings
In this family (Figure 1, Table 1), the proband, IV.2, was identified at 18 weeks gestation to have bilateral anophthalmia on fetal anomaly scan. No other anomalies were detected on the fetal scan and the karyotype was normal, 46,XY. The pregnancy was continued. Soon after birth, clinical anophthalmia was diagnosed and a CT scan revealed bilateral severely hypoplastic optic globes with only small residual structures, bilateral small optic nerves and chiasm, and normal pituitary tissue. There were no other intracranial abnormalities. No other abnormalities were found on clinical examination and the genitalia were normal. At the age of 2 years his developmental milestones were normal taking into account the visual impairment. His growth was also normal. The proband's mother (patient III.1) had bilateral iris and chorioretinal colobomata (Figure 1b), and her visual acuity was normal. There was a maternal family history of ocular abnormality with the proband's half great aunt (Patient II.5) and half great uncle (Patient II.9) both having microphthalmia with the right eye severely affected in both cases and a subtle iris pupillary defect in the left eye of patient II.9 (Figure 1b). Ophthalmic examination revealed ocular anomalies in other family members that were not known earlier. The proband's maternal grandmother (patient II.1) had thinning of the iris stroma inferiorly in the left eye (Figure 1b), whereas his maternal aunt (patient III.3) had a small retinal tuft inferiorly in the left eye (Figure 1b). His maternal great-grandmother (patient I.2) had microphthalmia and posterior embryotoxon bilaterally (Figure 1b) and a maternal half great aunt (patient II.4) had microcornea. Some individuals had hypermetropia (patients I.2, II.1, III.3 and IV.1) and some astigmatism (patients I.2 and III.3). Affected individuals in the pedigree were otherwise well. Other available individuals had normal ophthalmic examinations (patients II.2, II.3, III.2, III.4, III.5 and III.6).
SOX2 mutation in this four-generation family
We identified a novel mutation in the SOX2 gene in II.9 in this four-generation family. Analysis of genomic DNA revealed an A to G transition at nucleotide position 368 of SOX2 (c.368A>G) (Figure 2a). This mutation substitutes a highly conserved aspartic acid with glycine at amino acid position 123 (p.Asp123Gly) in the partner-factor interaction region immediately C-terminal to the HMG domain of SOX2 (Figure 2b).12 Sequencing in other family members revealed co-segregation of the mutation in individuals with ocular disease (Supplementary Figure 3, Table 1, Figure 1). The p.Asp123Gly mutation is not a known common variation (db SNP Build 129, http://genome.ucsc.edu/) and was not present in 254 control human chromosomes from Australians of UK descent. We examined the coding sequence and adjacent intronic regions of PAX6, CHX10, RAX and OTX2 for variations in patients II.5, II.9, III.1 and IV.2, and no pathogenic variations were identified.

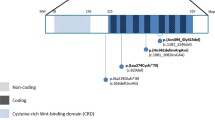
Schematic representation of B group SOX proteins and Sox2 expression. (a) (i) Direct sequencing of PCR product from affected patient II.9 in the family. Heterozygous mutation, an A to G transition at nucleotide 368 (NM_003106), which results in an aspartic acid to glycine substitution at amino acid 123 of the protein product. (ii) Control sequence. (b) Functional B group SOX domains including N-terminal domain, HMG DNA-binding and bending domain, group B homology partner-factor interaction region and C-terminal activation or repression domain.11 (c) Sequence conservation of the partner-factor interaction domain amongst the B group SOX proteins (SOX2, SOX1, SOX3, SOX14 and SOX21) and across species. The D123 position (indicated by an asterisk) is conserved in all known B group SOX proteins. Identical residues are shown in upper case, residues in the same polarity group in lower case letters and differences are dashed. (d) Parasagittal sections of embryonic mouse eyes examined for Sox2 expression. Signal is present in the neural retina, including the optic fissure margins, (i) just before closure of the optic fissure (arrow) at embryonic day 10.5 (E10.5) and (ii) during closure at E11.5.
Asp123 of the SOX2 protein is part of the B homology group region of the C-terminal domain of the B group SOX proteins. This B group homology region is critical for partner-factor binding. This aspartic acid residue is conserved in all known B group SOX proteins (Figure 2c). Replacement of this hydrophilic acidic residue with a hydrophobic glycine residue is expected to lead to abnormality in partner-factor binding.
Sox2 expression during optic fissure closure
Sox2 is expressed in the neuroectoderm of the head and the developing optic vesicle at E8 to E9.5 in the mouse.16 It is upregulated in the presumptive lens ectoderm at E9.5 and continues to be expressed in the lens and neural retina in early eye development.17 At E10.5 and E11.5, Sox2 expression is present at the optic fissure margins during optic fissure closure as shown in parasagittal sections (Figure 2d).
Discussion
We report a SOX2 mutation segregating in a multigenerational family. Family members have marked ocular phenotypic variability with features including bilateral clinical anophthalmia, bilateral optic fissure colobomata, unilateral inferior iris hypoplasia, microcornea and hypermetropia. The mutation results in the substitution of a conserved residue of the partner-factor-binding region of SOX2 and is absent from 254 control chromosomes. The findings in this family emphasize the importance of examination for SOX2 mutation in individuals with ocular defects such as optic fissure colobomata even in the absence of severe microphthalmia or anophthalmia.
The majority of SOX2 mutations have been identified in individuals with anophthalmia or severe microphthalmia. Most of the mutations cause premature termination codons because of nonsense or frameshift mutations, and whole gene deletions are also described.8 One frameshift mutation causing continued translation beyond the normal stop codon, and three missense mutations in the HMG domain have also been reported and all are associated with anophthalmia or severe microphthalmia.9, 11, 13 In this family, one patient has bilateral typical optic fissure closure defects (Figure 1, III.1) and two patients have iris anomalies indicating a minor form of optic fissure closure abnormality (Figure 1, II.1 and II.9). Typical optic fissure closure defects are not usually associated with SOX2 mutations. In the patient groups reported, there are only a few cases in which colobomata have been described and in most of these they are not reported to be typical optic fissure closure defect colobomata.8, 9, 18 More usual associated ocular features when present are cataract and the more severe ocular anomaly of sclerocornea.1, 8, 9 It is possible that severe abnormalities, such as anophthalmia, severe microphthalmia or marked abnormality of the early lens and anterior segment, are caused by loss-of-function mutations such as truncating, frameshift and HMG domain mutations that primarily affect SOX2 function in very early eye development. One report of a typical optic fissure closure defect and mild microphthalmia has been associated with a missense mutation in the transactivation domain of SOX2.19
The mutation described in the present family causes an amino acid change in a conserved region important in contributing to the tissue-specificity of SOX2 function (Figure 2b). SOX transcription factors exert tissue-specific effects in concert with tissue-specific partner factors. Partnership of SOX2 with Oct3 regulates expression of UTF1, an ES cell-specific coactivator, and partner-factor interaction contributes to SOX9 specificity in cartilage and genital ridge tissues.20, 21 In the lens, SOX2 interacts with the lens-specific factor δEF3, both of which bind to the DC5 enhancer and direct δ-crystallin lens-specific gene expression.20 The interaction between SOX2 and δEF3 is dependent on the conserved B group homology region immediately C-terminal to the HMG domain. The SOX2 mutation identified in this family, c.368A>G, p.Asp123Gly, leads to an amino acid change from a hydrophilic aspartic acidic residue to a hydrophobic glycine residue in this highly conserved B group partner-factor interaction region (Figure 2c). This may disrupt the binding of a SOX2 partner-factor, which is important in closure of the optic fissure.
The majority of earlier reported SOX2 mutations are de novo mutations.1, 8, 9 In view of this, it has been thought that SOX2 mutations may lead to reduced genetic fitness.8 The family we report is multigenerational and there have been no fertility problems. In addition, a number of extraocular features have earlier been described in association with SOX2 mutations, including poor growth, developmental delay, seizures, sensorineural hearing loss, esophageal abnormalities and male genital abnormalities. None of these are present in our family. Combinations of ocular and extraocular features may be dependent on the presence of a whole protein loss-of-function mutation. Mutations affecting the partner-factor interaction region as in the present family may lead to more specific ocular effects, and such mutations are more likely to contribute to multigenerational pedigrees.
SOX2 mutations are known to be associated with variability in the extraocular phenotype8 and non-penetrance is reported in one case.14 In view of the variable ocular phenotype in the present family, we considered the possibility that mutation in another gene/s, which may affect ocular development may be contributing to the ocular phenotype in the more severely affected individuals in this pedigree. Our investigation for mutations in OTX2, PAX6, RAX and CHX10 in the more severely affected individuals in this pedigree did not reveal any contributory pathogenic mutations. The discordant findings in the ocular phenotypes of monozygotic twins with a c.70del20 SOX2 mutation22 indicate that environmental or epigenetic factors may be contributing to the variable penetrance and expressivity associated with SOX2 mutations.
In conclusion, we report the first multigenerational family with a SOX2 mutation and the first SOX2 mutation, which lies in the highly conserved partner-factor interaction region of SOX2. Examination of a variety of eye phenotype patients for SOX2 mutations is useful in elucidating the role of SOX2 and its interacting partners in region and cell-specific differentiation of the eye.
Accession codes
References
Fantes J, Ragge NK, Lynch SA et al: Mutations in SOX2 cause anophthalmia. Nat Genet 2003; 33: 461–463.
Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL : PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet 1994; 7: 463–471.
Ragge NK, Brown AG, Poloschek CM et al: Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet 2005; 76: 1008–1022.
Voronina VA, Kozhemyakina EA, O'Kernick CM et al: Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum Mol Genet 2004; 13: 315–322.
Percin EF, Ploder LA, Yu JJ et al: Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet 2000; 25: 397–401.
Pasutto F, Sticht H, Hammersen G et al: Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet 2007; 80: 550–560.
Morrison D, FitzPatrick D, Hanson I et al: National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet 2002; 39: 16–22.
Bakrania P, Robinson DO, Bunyan DJ et al: SOX2 anophthalmia syndrome: 12 new cases demonstrating broader phenotype and high frequency of large gene deletions. Br J Ophthalmol 2007; 91: 1471–1476.
Ragge NK, Lorenz B, Schneider A et al: SOX2 anophthalmia syndrome. Am J Med Genet A 2005; 135: 1–7; discussion 8.
Chassaing N, Gilbert-Dussardier B, Nicot F et al: Germinal mosaicism and familial recurrence of a SOX2 mutation with highly variable phenotypic expression extending from AEG syndrome to absence of ocular involvement. Am J Med Genet A 2007; 143: 289–291.
Williamson KA, Hever AM, Rainger J et al: Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum Mol Genet 2006; 15: 1413–1422.
Kamachi Y, Uchikawa M, Kondoh H : Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet 2000; 16: 182–187.
Faivre L, Williamson KA, Faber V et al: Recurrence of SOX2 anophthalmia syndrome with gonosomal mosaicism in a phenotypically normal mother. Am J Med Genet A 2006; 140: 636–639.
Zhou J, Kherani F, Bardakjian TM et al: Identification of novel mutations and sequence variants in the SOX2 and CHX10 genes in patients with anophthalmia/microphthalmia. Mol Vis 2008; 14: 583–592.
Myat A, Henrique D, Ish-Horowicz D, Lewis J : A chick homologue of Serrate and its relationship with Notch and Delta homologues during central neurogenesis. Dev Biol 1996; 174: 233–247.
Wood HB, Episkopou V : Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech Dev 1999; 86: 197–201.
Furuta Y, Hogan BL : BMP4 is essential for lens induction in the mouse embryo. Genes Dev 1998; 12: 3764–3775.
Kelberman D, Rizzoti K, Avilion A et al: Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest 2006; 116: 2442–2455.
Wang P, Liang X, Yi J, Zhang Q : Novel SOX2 mutation associated with ocular coloboma in a Chinese family. Arch Ophthalmol 2008; 126: 709–713.
Kamachi Y, Cheah KS, Kondoh H : Mechanism of regulatory target selection by the SOX high-mobility-group domain proteins as revealed by comparison of SOX1/2/3 and SOX9. Mol Cell Biol 1999; 19: 107–120.
Nishimoto M, Fukushima A, Okuda A, Muramatsu M : The gene for the embryonic stem cell coactivator UTF1 carries a regulatory element which selectively interacts with a complex composed of Oct-3/4 and Sox-2. Mol Cell Biol 1999; 19: 5453–5465.
Zenteno JC, Perez-Cano HJ, Aguinaga M : Anophthalmia-esophageal atresia syndrome caused by an SOX2 gene deletion in monozygotic twin brothers with markedly discordant phenotypes. Am J Med Genet A 2006; 140: 1899–1903.
Acknowledgements
We thank Meredith Wilson and John Christodoulou for comments on the paper. We thank the family for their interest and participation in the research. Dr Ashley Seawright is acknowledged for clinical photography in one patient. We acknowledge support from the Peden and Thiess families and the Ophthalmic Research Institute of Australia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Mihelec, M., Abraham, P., Gibson, K. et al. Novel SOX2 partner-factor domain mutation in a four-generation family. Eur J Hum Genet 17, 1417–1422 (2009). https://doi.org/10.1038/ejhg.2009.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.79
Keywords
This article is cited by
-
Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia
Human Genetics (2019)
-
Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1
European Journal of Human Genetics (2014)
-
Isolated hypogonadotropic hypogonadism with SOX2 mutation and anophthalmia/microphthalmia in offspring
European Journal of Human Genetics (2011)
