
Abstract
We report maternal uniparental disomy of chromosome 17 (mat UPD17) in a 2.5-year-old girl presenting infantile cystinosis. This patient was homozygous for the 57 kb deletion encompassing the CTNS gene, frequently found in patients from the European origin. The proband's mother was heterozygous for the deletion and the father did not carry the deletion. We carried out haplotype analysis with polymorphic markers spanning the whole chromosome 17. Informative markers showed the presence of two maternal alleles but no paternal allele for regions spanning the 17q arm and the proximal half of 17p, and only one maternal allele on the distal 17p arm. As deletion of half of 17p is probably not viable, these results suggest mat UPD17 with heterodisomy of 17q and proximal 17p and isodisomy of distal 17p. This is the first demonstration of mat UPD17, in particular of isodisomy 17p, in cystinosis.
Similar content being viewed by others


Introduction
Cystinosis is an autosomal recessive disorder. It is characterized by an accumulation of intra-lysosomal cystine caused by a defect in cystine transport across the lysosomal membrane.1 Cystinosis is the most common inherited cause of proximal renal tubular dysfunction (the renal Fanconi syndrome). Affected individuals typically present with severe fluid and electrolyte disturbance (polyuria and polydypsia, vomiting, poor growth and rickets) at age 6–12 months. Within the first 2 years of age, patients also develop a severe and painful photophobia because of corneal cystine crystals. Without specific treatment, patients develop progressive growth retardation, with end-stage renal failure by 10 years of age.2 After renal transplantation, cystine continues to accumulate in other organs, leading to a multisystem disease. Three clinical forms of cystinosis (infantile, juvenile and ocular cystinosis) have been distinguished, based on severity of symptoms and age of onset.
Through positional cloning, the cystinosis locus has been mapped to the short arm of chromosome 17.3 Earlier studies have also identified the causative gene CTNS and pathogenic mutations.4 The CTNS gene maps to 17p13.3 and spans 23 kb. It encodes a 367 amino-acid protein, the lysosomal cystine transporter, cystinosin.4, 5 CTNS mutations have been detected in individuals affected with all forms of cystinosis. The most common mutation is a recurrent large 57 kb deletion spanning exons 1–10, detected in either the homozygous or the heterozygous state in ∼60–70% of northern European patients.6
In this study, we report maternal disomy of chromosome 17 in a girl with typical infantile cystinosis. This study provides the first evidence of uniparental disomy (UPD) of the whole chromosome 17 in cystinosis.
Materials and methods
Patient
The proband was born from healthy non-consanguineous parents of European origin after a full-term pregnancy and normal delivery. There were no earlier miscarriages and the family history was unremarkable (with two healthy brothers, 7 and 11 years old). At the age of 2 years, the proband presented with polyuria, failure to thrive and rickets. She had no developmental delay or neurological symptoms. Blood and urinary analyses showed hyperaminoaciduria, glycosuria, acidosis and renal electrolyte loss, confirming renal Fanconi syndrome. Corneal crystals were observed by slit-lamp examination and diagnosis of cystinosis was confirmed by a high leukocyte cystine content, measured at 6.75 nmol half-cystine/mg protein (normal <0.2).
PCR detection of breakpoint fragment in the CTNS gene
Blood samples were obtained from the proband and her parents, after informed consent from the parents. Genomic DNA was isolated from EDTA-anticoagulated blood by standard procedures. Primer sequences spanning a 360 bp junction fragment generated by the 57 kb deletion breakpoints, with PCR conditions for exons 9–12 of the CTNS gene, are described elsewhere.6
Chromosome analyses
Metaphase spreads were prepared from phytohaemaglutinin-stimulated peripheral blood lymphocyte cultures using standard procedures of hypotonic treatment and methanol/acetic acid fixation (3:1). RHG and GTG banding were performed according to standard protocols.
Analysis of microsatellite markers
The sequences, number, location and genotypes of microsatellite markers used in this study are shown in Table 1; data were obtained from the UniSTS database (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unists). Primer sequences, PCR amplification, gel electrophoresis, fluorescent genotyping and data analysis are described elsewhere.6
Results
Identification of the 57 kb deletion in a family with infantile cystinosis
Proband and control DNA were tested by the rapid PCR assay with the breakpoint primer sets.6 No PCR product was obtained in the control (data not shown), whereas an amplification product for the 360-bp breakpoint fragment was obtained in the proband, indicating that the proband bears the 57-kb deletion (blackened circle, Figure 1). The proband had amplified product for exons 11 and 12, but not for exons 9 and 10 of the CTNS gene, suggesting that she was a homozygous carrier of the 57-kb deletion. To confirm that this deletion was present on both alleles of the patient, we investigated the segregation of the deletion in her parents. Her parents had amplified product for all exons tested. As expected, the 360-bp deletion fragment was present in her mother, indicating that the mother was a heterozygous carrier for the 57-kb deletion; unexpectedly, the deletion fragment was not found in her father (Figure 1).

Segregation of the 57 kb deletion in a family with nephropathic cystinosis. The proband (black circle) displayed amplified products for exons 11 and 12 of the CTNS gene and for the 360 bp breakpoint fragment corresponding to the 57 kb deletion. No amplified product was detected for exons 9 or 10 of the CTNS gene. Her mother displayed amplified products for all tested exons and for the 360 bp fragment, suggesting that she is heterozygous for the 57 kb deletion. Her father displayed amplified products for all tested exons, but not for the 360 bp fragment indicating that he does not display the 57 kb deletion.
Characterization of maternal UPD17
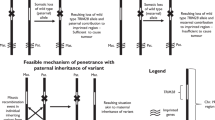
We tested four microsatellite markers, surrounding and within the 57-kb deletion (D17S831, D17S1798, D17S829 and D17S1828). Proband DNA was amplified for all markers surrounding the deletion, but not for the D17S829 marker located within the CTNS 57-kb deletion. This confirmed the homozygous status of a CTNS deletion in the patient (Table 1). All markers were amplified for both parents. No paternal markers were detected at the patient CTNS locus. Non paternity was excluded by use of seven microsatellite markers from six different chromosomes (DXS1073, D2S159, D4S395, D4S3038, D7S2563, D11S919 and D16S3124 – data not shown). Genotyping of 22 additional markers spanning the entire chromosome 17 in the family showed non-Mendelian segregation of all informative markers (D17S1866, D17S1529, D17S831, D17S829, D17S1852, D17S921, D17S1794, D17S1824, D17S1861, D17S791, D17S787 and D17S1830), with a complete absence of paternal contribution (Table 1). We found two maternal alleles for chromosome 17q and the proximal half of chromosome 17p, and reduction to homozygosity of the maternal alleles in the distal half of chromosome 17p, with a recombination event mapping between D17S1852 and D17S921. Routine chromosome analysis on the proband's peripheral blood lymphocytes showed a normal 46, XX karyotype. Thus, the abnormal genotype of the proband could result from the presence of two maternal alleles and no paternal allele for the informative markers spanning the 17q arm (D17S1824, D17S1861, D17S791, D17S787 and D17S1830) and only one maternal allele from the 17p arm, because of either a 17p13.3 deletion or to maternal uniparental isodisomy in this part of the genome.
Discussion
We report a patient with isolated cystinosis because of a homozygous deletion of 10 exons of the CTNS gene. This deletion is associated with an absence of paternal contribution over the entire chromosome 17 and reduction to homozygosity for maternal alleles in the distal half of chromosome 17p. This could be caused by paternal deletion at 17p13.3 or maternal uniparental isodisomy of chromosome 17 (mat UPD17). A 17p deletion would probably not be viable and was excluded both by normal karyotype analysis (data not shown) and the absence of any extra-renal or ophthalmic symptoms. In particular, paternal deletion of this region would encompass the LIS1 gene (involved in Miller–Dieker syndrome, MDS). The proband did not show MDS symptoms such as seizures or a typical EEG fast spike pattern during the first year of life, consistent with exclusion of a paternal deletion.
Uniparental disomy occurs when a child receives both copies of a particular chromosome (or part of a chromosome) from only one parent, thus distorting the concept of biparental inheritance. UPD may involve two copies of the same chromosome (isodisomy) or one copy from the contributing parent's chromosome pair (heterodisomy). Our results suggest mat UPD17 with heterodisomy of 17q and isodisomy of a large part of 17p.
Uniparental heterodisomy can arise from the fertilization of aneuploid gametes, followed by either gametic complementation or trisomic rescue. Meiotic recombination events can result in a mixed UPD with interspersed regions of heterodisomy and isodisomy along the chromosome.7 In our proband, the initial error could result from non-disjunction in meiosis I.
Uniparental disomy involving different chromosomes has been described in several cases of human disease. A review of the literature shows only one case of maternal UPD17 reported earlier.8, 9 This earlier study described maternal disomy of chromosome 17, with proximal (pericentromeric) homozygosity and distal heterozygosity in a boy whose history included an apparently normal pregnancy outcome and normal postnatal development.8 In our study, with the exception of renal signs and photophobia clearly related to cystinosis, no phenotypic abnormalities (dysmorphic features or multiple organ anomalies) were noted on physical examination of the proband. The phenotype of our patient was clearly the consequence of the CTNS gene deletion, (itself because of the disomy) and not of the maternal UPD17. In the absence of phenotypic abnormalities in these two children, these reports suggest that chromosome 17 is not likely to be subject to imprinting.8, 10, 11
Rio et al9 reported segmental maternal heterodisomy for a small 11-cM region of chromosome 17q in a child with behavioral disorder, severe mental retardation and facial dysmorphism. As chromosome 17 is unlikely to be subject to imprinting, a complementary CGH array study could be useful to search for additional deletions or duplications. In such cases, somatic events such as mitotic recombination (causing segmental UPDs) or duplication of a viable chromosome to compensate for an inherited dysfunctional chromosome can also result in UPD.7
Genetic counseling for the proband's parents will be different from that offered for a simple homozygous deletion without UPD17. UPD is a rare genetic event and the risk of recurrence is null almost. The risk that the father has a mutation in the CTNS gene is also very low, as he does not have the CTNS 57 kb deletion (which represents 75% of CTNS mutations in the European population). Thus, the risk of having another child affected with cystonosis is extremely low.
In conclusion, our report is the first to show maternal disomy of chromosome 17 in association with an isolated renal disorder. UPD17 does not seem to be associated with other abnormal phenotypes, suggesting that there are probably no imprinted genes on chromosome 17.
References
Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD : Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 1982; 217: 1263–1265.
Gahl WA, Ingelfinger J, Mohan P, Bernardini I, Hyman PE, Tangerman A : Intravenous cysteamine therapy for nephropathic cystinosis. Pediatr Res 1995; 38: 579–584.
The Cystinosis Collaborative Research Group: Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. Nat Genet 1995; 10: 246–248.
Town M, Jean G, Cherqui S et al: A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet 1998; 18: 319–324.
Kalatzis V, Cherqui S, Antignac C, Gasnier B : Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J 2001; 20: 5940–5949.
Forestier L, Jean G, Attard M et al: Molecular characterization of CTNS deletions in nephropathic cystinosis: development of a PCR-based detection assay. Am J Hum Genet 1999; 65: 353–359.
Bruce S, Leinonen R, Lindgren CM et al: Global analysis of uniparental disomy using high density genotyping arrays. J Med Genet 2005; 42: 847–851.
Genuardi M, Tozzi C, Pomponi MG et al: Mosaic trisomy 17 in amniocytes: phenotypic outcome, tissue distribution, and uniparental disomy studies. Eur J Hum Genet 1999; 7: 421–426.
Rio M, Ozilou C, Cormier-Daire V et al: Partial maternal heterodisomy of chromosome 17q25 in a case of severe mental retardation. Hum Genet 2001; 108: 511–515.
Martinet D, Vial Y, Thonney F, Beckmann JS, Meagher-Villemure K, Unger S : Fetus with two identical reciprocal translocations: description of a rare complication of consanguinity. Am J Med Genet A 2006; 140: 769–774.
Kotzot D, Utermann G : Uniparental disomy (UPD) other than 15: phenotypes and bibliography updated. Am J Med Genet A 2005; 136: 287–305.
Acknowledgements
We are grateful to the patient and her family for their cooperation. This study was supported by Vaincre les Maladies Lysosomales, the Cystinosis Research Foundation and the Cystinosis Research Network.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lebre, AS., Morinière, V., Dunand, O. et al. Maternal uniparental heterodisomy of chromosome 17 in a patient with nephropathic cystinosis. Eur J Hum Genet 17, 1019–1023 (2009). https://doi.org/10.1038/ejhg.2009.13
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.13
Keywords
This article is cited by
-
Clinical utility gene card for: Cystinosis
European Journal of Human Genetics (2014)
-
Maternal Uniparental Isodisomy Causing Autosomal Recessive GM1 Gangliosidosis: A Clinical Report
Journal of Genetic Counseling (2014)
-
Complete Paternal Isodisomy of Chromosome 17 in Junctional Epidermolysis Bullosa with Pyloric Atresia
Journal of Investigative Dermatology (2010)
