
Abstract
The cerebellum is the primary motor coordination centre of the central nervous system. Lesions or congenital defects of the cerebellum cause incoordination of the muscles resulting in irregular gait and falling. Recently, we reported a large family with cerebellum hypoplasia and quadrupedal locomotion as a recessive trait, which we mapped to chromosome 17p13. We identified one additional family with the same condition and mapped the underlying gene to a 14-cM interval on chromosome 9ptel using a genome-wide linkage approach. Sequencing of candidate genes identified a homozygous frameshift mutation in the very low-density lipoprotein receptor (VLDLR) gene in all affected individuals. The association of cerebellar hypoplasia with mutations in VLDLR has been reported previously in the Hutterite population and in a family from Iran. However, quadrupedal locomotion was never observed indicating that environmental factors play a major role in the pathogenesis of this form of locomotion.
Similar content being viewed by others


Introduction
The cerebellum is the control centre of the central nervous system for muscle tone, balance, and the coordination of movement. Numerous cerebellar malformations have been described in humans, which share the prototypic feature of impaired walking with a lack of coordination of gait and limbs. Recently, we reported a large Turkish family with five offsprings that have never learned to walk on two legs due to a severe combined cerebellar and extrapyramidal movement disorder.1, 2 Instead, they have fully adapted to quadrupedal palmigrade locomotion, a unique feature highlighted in a BBC documentary (‘The Family that Walks on All Fours’, 17 March 2006). This unique trait was mapped to chromosome 17p13 and was named ‘Cerebellum Hypoplasia, Mental Retardation and Quadrupedal Locomotion syndrome’ (MIM 610185). We have now studied a second Turkish family with the same clinical condition present in three close relatives (Figure 1).

Pedigree of family.
Materials and methods
Patient data
The index patient, a 38-year-old man, originates from a very small village in the west part of Turkey. He has one healthy brother. His sister died of an unknown reason at the age of 4 years. He was born after an uneventful gestation and delivery. He began to walk on all fours after the age of 2 years. He was never able to walk bipedally. On the basis of clinical assessment, he was found to be severely mentally retarded. The neurological examination revealed a severe degree of cerebellar ataxia, intentional tremor, dysarthria bilateral dysmetria, and dysdiadochokinesia. No pyramidal signs were found. His body length was below the third percentile, but head circumference was normal. The sister and brother of his father showed the same type of ataxia and the same gait phenotype as the proband. The sister (aunt of the index patient) is able to walk bipedally without assistance but prefers to use quadrupedal locomotion. The index patient lives with his family. His aunt and uncle live in another house by themselves, but the relatives support them in their daily work of life. The degree of their mental impairment is moderate to severe. They did not attend school, and their verbal communication is impaired. The lipid status of the affected individuals is not known.
A magnetic resonance analysis was obtained from the index patient.
Molecular analysis
After obtaining written informed consent, we took blood samples from available family members. DNA was extracted by standard protocols. A homozygosity for 17p13 was ruled out. A genome-wide scan using the 10 K Affymetrix SNP chip revealed a single locus for homozygosity on chromosome 9p between 0.00 and 14 cM. This interval includes an obvious candidate, very low-density lipoprotein receptor (VLDLR), which is part of the reelin (RELN) signalling pathway and is responsible for dysequilibrium syndrome (DES) in the Hutterite population. Focusing on VLDLR, we amplified the coding region of the VLDLR, composed of 19 exons. Standard protocol PCRs were carried out to amplify exons 1–19. PCR products were analysed by 2% agarose gel electrophoresis. Purified PCR products were sequenced in both directions by using PCR primers as sequencing primers and the Applied Biosystems Prism BigDye terminator cycle sequencing reaction kit. The products were evaluated on an Applied Biosystems 3100 DNA sequencer.
Results
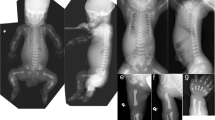
Magnetic resonance imaging (MRI) of the index patient revealed hypoplasia of the cerebellum and vermis as well as mild hypoplasia of the corpus callosum (Figure 2). Brainstem including midbrain and pons was of normal size. These MRI findings are similar to our first family described with cerebellar hypoplasia and quadrupedal locomotion.

(a) Three affected individuals showing quadrupedal locomotion. (b) MRI of brain showing hypoplasia of the cerebellum.
Sequencing of VLDLR revealed a homozygous frameshift mutation (c.2339delT, p.Ile779fsX3) in exon 17 of the VLDLR gene in all three of the affected members of the family (Figure 3). This mutation results in a premature stop codon after two additional amino acids. VLDLR has 19 exons. Exon 17 codes for amino acids 781–806 and as such contains part of the transmembrane helices (amino acids 797–819).

(a) Exon stucture of VLDLR and localisation of mutation. (b) Electropherogram showing mutation.
Discussion
Dysequilibrium syndrome is an autosomal recessive condition characterised by non-progressive cerebellar ataxia, moderate-to-profound mental retardation, delayed ambulation, and a static clinical course. The designation ‘Dysequilibrium syndrome’ was given3, 4 to a form of cerebral palsy with autosomal recessive inheritance characterised by a variety of congenital abnormalities, including mental retardation in most cases, disturbed equilibrium with severely retarded motor development, muscular hypotonia, and perceptual abnormalities indicative of widespread brain dysfunction. Nine years later, Schurig et al5 presented 11 new cases among the Dariusleut Hutterites of Alberta. The patients presented with hypotonia at birth, ataxia, as well as motor and mental retardation, which became apparent in the first year of life. Consistent signs were unsteady, broadly based gait and stance, exaggerated deep tendon reflexes mainly in the lower limbs, and mild-to-moderate mental retardation. All affected showed late ambulation (after age 6 years), but quadrupedal locomotion was never observed.6 Computerised axial tomography showed cerebellar atrophy. Pallister and Opitz7 observed the same condition in the Dariusleut Hutterites of Montana. Using an identity-by-descent mapping approach, Boycott et al8 localised the gene for this syndrome to chromosome region 9p24 and identified a 199-kb homozygous deletion encompassing the entire VLDLR gene in all affected individuals.
Recently, a homozygous nonsense mutation (c.1342C>T, R448X) in exon 10 of the VLDLR gene in an Iranian Family with DES was reported. The affected individuals could not walk independently as described by Hutterites patients; however, quadrupedal locomotion has not been observed.8 Both reported mutations can be expected to result in loss of the VLDLR protein. The Hutterine mutation is a complete deletion of the gene and as such does not result in a gene product. The Iranian mutation results in a premature stop codon approximately in the middle of the mRNA, truncating the indespensable transmembrane domain (amino acids 797–819) and half of the receptor domain from the N terminus of the protein. In addition, this mutation is likely to result in nonsense-mediated decay of the mRNA. Thus, the mutation can be expected to cause a complete loss of function. The mutation reported here results in a premature stop codon in exon 17 at amino-acid position 779. Similar to the Iranian mutation, this change is likely to result in nonsense-mediated decay. Amino acid 779 is N-terminal of the transmembrane domain. Accordingly, this mutation also results in a truncated protein without transmembrane domain, if translated. Thus, the overall effect of the three reported mutations is likely to be similar, that is, a loss of VLDLR protein.
The VLDLR belongs to the mammalian LDL receptor (LDLR) gene family. The LDLR gene family comprises a group of structurally related multifunctional cell-surface receptors (VLDLR, apolipoprotein E receptor 2 (ApoER2), the LDL receptor, LRP and Megalin) that mediates endocytosis of extracellular ligands.9 All members are characterised by common structural features that include (1) cysteine-rich repeats consisting of 40 amino-acid residues in the ligand-binding domain or in the complement-type domain; (2) epidermal growth factor precursor-type repeats; (3) module of 50 amino-acid residues with a consensus tetrapeptide, YWTD; (4) a single transmembrane domain; and (5) a cytoplasmic domain containing an NPXY sequence required for clustering of the receptor into coated pits.10
VLDLR is part of the RELN signalling pathway, which guides neuroblast migration in the cerebral cortex and cerebellum. Migration of neuroblasts and neurons is an essential process in the development of the nervous system. RELN, an extracellular matrix protein secreted by CAjalRetzius cells in the developing marginal zone of the cortex and other laminated brain regions, is necessary for an early, specific step in the migration of several hindbrain nuclei.11, 12, 13 It directs the organisation of cortical structures of the brain by clustering its receptors, the VLDLR and the ApoER2, causing the activation of src-family kinases (SRKs) and the phosphorylation of the adapter molecule, the Disabled-1Dab1.14 RELN may act as a stop signal,15 a chemoattractant,16 and a detachment signal for migrating neurons.17, 18
Disruption of RELN in homozygous reeler mice results in failed migration of the Purkinje cells, cortical layer disruption, cerebellar hypoplasia, and ataxia.19 Human homozygous RELN mutations are associated with an autosomal recessive lissencephaly with severe cerebellar hypoplasia. Clinically, patients have developmental delay, hypotonia, severe ataxia, and seizures, whereas brain MRI demonstrates diffuse pachygyria, hippocampal dysplasia, and a profoundly hypoplastic cerebellum and brainstem, with a nearly complete absence of cerebellar folia.20
Vldlr knockout mice appear grossly and neurologically normal and have a normal life span.21 However, the phenotype of the Vldlr defect manifests itself mainly in the cerebellum. In the Vldlr−/− cerebellum, Purkinje cells are ectopically located, apparently due to their inability to properly respond to the RELN signal that emanates from the granule cells in the external granular layer. In the cortex, Vldlr and mDab1 are selectively expressed in the migrating neurons that are about to make contact with RELN in the marginal zone, consistent with a role of Vldlr as a receptor for a migratory stop signal. In the Vldlr-deficient cortex, neurons are strictly radially aligned, and although they apparently have reached their normal assigned layer, they have failed to distribute within that layer.22
Although dysequilibrium patients typically learn to walk very late (between the age of 5 and 21 years) or else never walk at all, the individuals of the family presented here differ from the DES patients by adopting complete quadrupedal locomotion. As in the case of the quadrupedal individuals we reported previously, they step with their hands and feet in both diagonal and lateral sequence gaits. The hands are placed down, with the weight taken on the wrists and lower ulnar area of the palm. They walk with fully stretched knee and elbow joints. Only one patient can walk bipedally without assistance; however, she mostly prefers walking on all fours. Quadrupedal locomotion in these individuals appears to be an adaptation for dealing with their ataxia. But such an adaptation is unusual, as human skeletons have evolved over four million years for walking on two legs.
This is the first report of a VLDLR mutation associated with human quadrupedal locomotion and cerebellum hypoplasia. The switch from quadrupedal locomotion to bipedalism is a fundamental adaptation in human evolution that sets hominins apart from all other primates. Clearly, many morphological changes were necessary for this complex transition, involving adaptive changes in the skeletal and the central nervous system. However, little is known about the genes and the genetic evolution that ultimately resulted in the full adaptation of human ancestors to bipedalism. The family presented here shows us that although the morphological adaptive changes for bipedalism in the skeletal and central nervous systems have been established, efficient quadrupedal locomotion is still possible in humans. The absence of quadrupedal locomotion in the Hutterite and Iranian patients with DES indicates that similar gene defects, that is inactivation of the VLDLR gene product, result in similar morphological outcome, that is cerebellar hypoplasia, but different ways of compensation. Although the Hutterite and Iranian patients did not learn to walk before the age of 6 years or never, the affected individuals described here and in our earlier report continued to crawl and adapted a childhood bear walk for rapid and efficient movement. It may well be that this form of movement was not considered appropriate in the Hutterite community and was therefore suppressed, although why it was tolerated in these two Turkish families remains unknown. We conclude that although the precondition for quadrupedal locomotion is cerebellar hypoplasia and subsequent ataxia, the emergence of this trait as a behavioral phenotype depends on special environmental influences during child development.
References
Turkmen S, Demirhan O, Hoffmann K et al: Cerebellar hypoplasia and quadrupedal locomotion in humans as a recessive trait mapping to chromosome 17p. J Med Genet 2006; 43: 461–464.
Humphrey N, Skoyles JR, Keynes R : Human hand-walkers: five siblingswho never stood up. LSE 2005 CPNSS Discussion Paper, DP 77/05).
Hagberg B, Sanner G, Steen M : The dysequilibrium syndrome in cerebral palsy. Clinical aspects and treatment. Acta Paediatr Scand Suppl 1972; 226: 1–63.
Sanner G : The dysequilibrium syndrome. A genetic study. Neuropadiatrie 1973; 4: 403–413.
Schurig V, Orman AV, Bowen P : Nonprogressive cerebellar disorder with mental retardation and autosomal recessive inheritance in Hutterites. Am J Med Genet 1981; 9: 43–53.
Glass HC, Boycott KM, Adams C et al: Autosomal recessive cerebellar hypoplasia in the Hutterite population. Dev Med Child Neurol 2005; 47: 691–695.
Pallister PD, Opitz JM : Disequilibrium syndrome in Montana Hutterites. Am J Med Genet 1985; 22: 567–569.
Boycott KM, Flavelle S, Bureau A et al: Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet 2005; 77: 477–483.
Moheb LA, Tzschach A, Garshasbi M et al: Identification of a nonsense mutation in the very low-density lipoprotein receptor gene (VLDLR) in an Iranian family with dysequilibrium syndrome. Eur J Hum Genet 2008; 16: 270–273.
Krieger M, Herz J : Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP). Annu Rev Biochem 1994; 63: 601–637.
Tiebel O, Oka K, Robinson K et al: Mouse very low-density lipoprotein receptor (VLDLR): gene structure, tissue-specific expression and dietary and developmental regulation. Atherosclerosis 1999; 145: 239–251.
Rossel M, Loulier K, Feuillet C, Alonso S, Carroll P : Reelin signaling is necessary for a specific step in the migration of hindbrain efferent neurons. Development 2005; 132: 1175–1185.
Hack I, Hellwig S, Junghans D et al: Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development 2007; 134: 3883–3891.
D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T : Reelin is a ligand for lipoprotein receptors. Neuron 1999; 24: 471–479.
Zhang G, Assadi AH, McNeil RS et al: The Pafah1b complex interacts with the Reelin receptor VLDLR. PLoS ONE 2007; 2: e252.
Dulabon L, Olson EC, Taglienti MG et al: Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron 2000; 27: 33–44.
Gilmore EC, Herrup K : Cortical development: receiving reelin. Curr Biol 2000; 10: R162–R166.
Hack I, Bancila M, Loulier K, Carroll P, Cremer H : Reelin is a detachment signal in tangential chain-migration during postnatal neurogenesis. Nat Neurosci 2002; 5: 939–945.
Sanada K, Gupta A, Tsai LH : Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron 2004; 42: 197–211.
Rice DS, Sheldon M, D’Arcangelo G, Nakajima K, Goldowitz D, Curran T : Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 1998; 125: 3719–3729.
Hong SE, Shugart YY, Huang DT et al: Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 2000; 26: 93–96.
Frykman PK, Brown MS, Yamamoto T, Goldstein JL, Herz J : Normal plasma lipoproteins and fertility in gene-targeted mice homozygous for a disruption in the gene encoding very low density lipoprotein receptor. Proc Natl Acad Sci USA 1995; 92: 8453–8457.
Trommsdorff M, Gotthardt M, Hiesberger T et al: Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999; 97: 689–701.
Acknowledgements
We would like to thank the family for participation in this study. We are grateful to Arlene Carol, who first brought the case to our attention.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Türkmen, S., Hoffmann, K., Demirhan, O. et al. Cerebellar hypoplasia, with quadrupedal locomotion, caused by mutations in the very low-density lipoprotein receptor gene. Eur J Hum Genet 16, 1070–1074 (2008). https://doi.org/10.1038/ejhg.2008.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.73
Keywords
This article is cited by
-
Very mild features of dysequilibrium syndrome associated with a novel VLDLR missense mutation
neurogenetics (2016)
-
Cerebellar ataxia, mental retardation and dysequilibrium syndrome 1 (CAMRQ1) caused by an unusual constellation of VLDLR mutation
Journal of Neurology (2013)
-
A missense founder mutation in VLDLR is associated with Dysequilibrium Syndrome without quadrupedal locomotion
BMC Medical Genetics (2012)
-
Macrophage VLDL receptor promotes PAFAH secretion in mother's milk and suppresses systemic inflammation in nursing neonates
Nature Communications (2012)
-
Novel VLDLR microdeletion identified in two Turkish siblings with pachygyria and pontocerebellar atrophy
neurogenetics (2010)
