
Abstract
The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Phase 1 Schizophrenia trial compared the effectiveness of one typical and four atypical antipsychotic medications. Although trials such as CATIE present important opportunities for pharmacogenetics research, the very richness of the clinical data presents challenges for statistical interpretation, and in particular the risk that data mining will lead to false-positive discoveries. For this reason, it is both misleading and unhelpful to perpetuate the current practice of reporting association results for these trials one gene at a time, ignoring the fact that multiple gene-by-phenotype tests are being carried out on the same data set. On the other hand, suggestive associations in such trials may lead to new hypotheses that can be tested through both replication efforts and biological experimentation. The appropriate handling of these forms of data therefore requires dissemination of association statistics without undue emphasis on select findings. Here we attempt to illustrate this approach by presenting association statistics for 2769 polymorphisms in 118 candidate genes evaluated for 21 pharmacogenetic phenotypes. On current evidence it is impossible to know which of these associations may be real, although in total they form a valuable resource that is immediately available to the scientific community.
Similar content being viewed by others

Introduction
One notable result of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) trial of antipsychotic effectiveness in schizophrenia1 was the high discontinuation rates among the four atypical and one typical antipsychotic drugs trialed – ranging from 64% for olanzapine to 82% for quetiapine. An even more striking outcome was the rough equivalence among the medicines in most measures when patients on each drug are considered as a group. Although current medicines therefore do not appear better or worse on average, at the individual patient level, variation in response is pronounced. A major goal of psychiatric pharmacogenetics is therefore the prediction of the drug that will work best, with the fewest side effects, for individual patients with schizophrenia.
Unfortunately, our capacity to predict therapeutic response and clinically significant side effects in individual patients is currently extremely limited. There are, for example, no effective means by which to match individual patients to medications that offer them more symptom control or that reduce their chances or severity of adverse reactions. In general, studies implicating genetic variants in antipsychotic response have not been well replicated, with follow-up studies often using different genetic markers and models, testing heterogeneous study populations, and affected by a publication bias of positive findings.2, 3 There may be some cumulative evidence for associations with weight gain and tardive dyskinesia, although effect sizes are generally small.3, 4, 5, 6 There are examples of genetic associations with antipsychotic response that have led to the development of predictive tests: a human leukocyte antigen test that measures the risk of developing agranulocytosis when treated with clozapine,7 and a combination of polymorphisms that can be used in certain patient groups to predict clozapine response;8 however, both these tests are of very limited clinical utility.5 Finally, the AmpliChip CYP450 pharmacogenetic test was hoped to help predict antipsychotic response.9 However, a recent study showed that these genotypes cannot predict antipsychotic dose or response in real-world settings,10 and the AmpliChip too is expected to be of little utility in antipsychotic prescribing.11
Here we have identified 21 response phenotypes, some of which are partially correlated, and evaluated them in the 756 CATIE participants consented for genetic studies. Although in the CATIE trial, the primary phenotype was discontinuation of treatment for any cause, we felt that this end point was too heterogeneous for a genetic study. Instead, we examined CATIE secondary outcome measures, including discontinuation due to inefficacy, weight gain and change in PANSS scores. To reduce the scope for data mining, our philosophy for defining these pharmacogenetic phenotypes was, wherever possible, to follow how CATIE investigators evaluated and reported responses in the trial. For example, a weight-gain ‘case’ was defined as a patient who gained 7% or more of his or her baseline body weight (cf. Lieberman et al.1). Finally, as neurocognitive impairment in schizophrenia is a significant predictor of outcome12 and has been suggested to be a particularly suitable trait for the genetic study of schizophrenia,13 we searched for genetic predictors of cognitive change, utilizing data from detailed cognitive assessments performed throughout the CATIE trial and reflecting various measures of verbal learning and memory, working memory, motor function, attention and executive function. All phenotypes in this paper were defined before genetic analyses were performed to prevent false positives due to unconstrained or biased data mining.
Materials and methods
A detailed description of the participants consented for genetic study is provided in Sullivan et al.14 SNPs were genotyped using Illumina GoldenGate technology. For further description of the CATIE trial and participants and detailed description of gene and SNP selection, genotyping and quality control, phenotype definitions and statistical analyses, please see Supplementary Methods. All genes examined and their SNP coverage are shown in Table 1; all phenotypes and sample sizes for each association test are shown in Table 2.
Results
In total, 21 phenotypes were tested (Table 2). Two associations were significant after permutation correction for all tested SNPs (see below). No associations were significant after correction for all SNPs and all phenotypes tested (P<9 × 10−7).
Neurocognition
Study-wide significant association
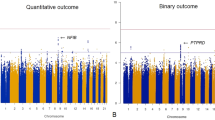
SNP rs7778604, located in intron 2 of the GRM8 gene, was associated with antipsychotic-induced change in verbal memory, and this remained significant after permutation correcting for all studied SNPs (corrected P=0.02, uncorrected P=0.00001, n=447; Figure 1). The SNP was present in both African-American and European-American participants, and to ensure that the association was not caused by uncorrected stratification, we repeated the analysis of this phenotype with 10 axes of ancestry, instead of a single axis. The association remained significant at 0.00037 (uncorrected), but was no longer significant after correction by permutation for all tested SNPs (P=0.58). Those patients carrying the rarer variant (n=97) tended to improve in verbal memory with 8 weeks of treatment, whereas those homozygous for the common allele (n=369) performed worse after 8 weeks of phase 1 treatment. The associated SNP is in a highly alternatively spliced gene but has no obvious function. It is in strong linkage disequilibrium (LD) with a number of other intronic SNPs, including rs17863182, which lies in the middle of a highly conserved region 3 kb upstream from another alternative exon.

Association of GRM8 SNP rs7778604 with change in verbal memory (n=447; uncorrected P=0.00001). On average, the patients with the less common allele showed an improvement in memory, whereas the patients homozygous for the common allele did not.
Multiple hits
To identify SNPs with a general effect across cognitive domains, we first looked at the top hits associated with the principal component axis drawn from the change measures of all tests. The strongest association was with rs10954143, an intronic SNP in GRM8 (P=0.0003) that was associated with many other intronic SNPs. This association seemed to be primarily driven by letter-number sequencing (P=0.001), continuous performance test (0.002) and mazes (P=0.03), so it is associated with a number of different domains. Similarly, the next strongest associated SNPs were two tightly linked SNPs in the 3′-UTR of ADCY1, rs2461127 and rs2471267 (each with P=0.0005), and these were also driven by associations in letter-number sequencing (P=0.01) and continuous performance (P=0.06), as well as verbal recall (P=0.01) and FAS (P=0.06).
We then looked at the uncorrected P-values to examine all hits in common across multiple cognitive tests (<0.05), not including PC1 (Table 3). No SNPs were associated with all nine tests. The SNP that was associated with most tests (5/9) was rs1011427, an intronic SNP in HTR4, and was in complete LD with another intronic SNP. HTR4 codes for the serotonin receptor 4 subunit, and has been associated with enhanced cognition in animals.15, 16, 17 This SNP was associated with verbal fluency categories and letters, visuospatial working memory, verbal recall and digit symbol score (Table 3). As expected, it was also associated with the PC1 measure (P=0.006).
Other SNPs of particular interest include an intronic SNP in ADCY1, rs11766222 (PC1, P=0.002). ADCY1 codes for the neurospecific type I adenylyl cyclase, involved in hippocampal long-term potentiation and certain forms of learning and memory. This SNP is in complete LD (r2=1) with several other SNPs surrounding exons 12, 13 and 14, and associates with verbal fluency (letters), the auditory letter-number test, the continuous performance test and mazes. Also, of interest was rs1390938 (The136Ile), a non-synonymous coding SNP in SLC18A1, the gene that codes for the vesicular monoamine transporter 1 (PC1, P=0.008). This SNP has previously been associated with bipolar disorder18 and in this study associated with verbal fluency (categories), the auditory letter-number test, verbal recall and digit symbol. All SNPs that associated with three or more of the cognitive phenotypes are displayed in Table 3.
Top hits
We next examined the strongest cognitive associations. The lowest P-value was for rs17866959, an intronic SNP in GRM8 that was present only in African-American patients and associated with change in Wisconsin card sorting test (WCST) score. The P-value for association with change in WCST errors was P=8.3 × 10−6, and considering only African-Americans, the P-value was 2.4 × 10−6. Those patients carrying the variant allele all declined in WCST performance except one participant whose score did not change; however, those participants not carrying this variant tended to slightly improve in performance with treatment.
The next two strongest hits, and the only others with P<10−4, were both intronic SNPs in GRM7 that were associated with a change in visuospatial memory. The SNPs are rs7627369 and rs3864075, and they are in partial LD with each other (r2=0.25 in this data set). Those individuals, both African-American and European-American, that carried the rare variant showed a stronger improvement in score across the 8 weeks of treatment (eg, rs7627369 homozygotes carrying the common variant made an average of 0.07 fewer errors versus those carrying the rare variant, who made 0.90 fewer errors after 8 weeks of treatment). SNP rs7627396 also associates with change in PANSS negative (P=0.0002) and PANSS general psychopathology (P=0.0004) scores.
We annotated the list of cognitive hits (<0.05) using WGAviewer,19 and some of the SNPs seemed to be of particular note due to their position or the position of associated SNPs. SNPs that had a P-value <0.001, and also SNPs that had a P-value <0.01 and were located in a coding or otherwise putatively functional region of the gene, are displayed in Table 4. All SNPs with a P-value of association with a cognitive phenotype <0.05, along with their annotation, are displayed in Supplementary Results Table S1.
PANSS
No PANSS association stood up to correction for all tested SNPs. All P-values described are uncorrected.
Multiple hits
To see if any SNP is associated with an overall improvement in symptoms, we looked first at SNPs that associated with PANSS total, and thus associated with multiple PANSS measures. Table 5 shows all 22 SNPS that are associated with PANSS total score with an uncorrected P-value of <0.01, along with their P-value for association with each of the three individual PANSS scores. The strongest PANSS total association was rs7627369, an intronic SNP in the metabotropic glutamate receptor gene GRM7 that is of low frequency (4%) in both European- and African-Americans (and is also associated with visuospatial working memory, above). It is located in intron 8 of the gene and is associated with other SNPs in introns 8 and 9. Altogether, there were 19 participants who carried the rare allele at this locus who also had change scores for PANSS available at 3 months on the phase I drug. For each PANSS category, those with the rare allele showed greater improvement in score across the 3 months. On average, patients carrying the rare allele dropped 17.3 PANSS total points in comparison with 6.5 points dropped by patients homozygous for the common allele.
SLC1A6, a glutamate transporter gene, showed three associated SNPs but only two of the SNPs were in LD, indicating two independent hits in the same gene. One of the associated SNPs, rs3746295, is a synonymous coding SNP in the first exon.
There was another SNP located in a coding region that was associated with PANSS score. This was rs4586, in exon 2 of CCL2 (aka SCYA2, MCP1 and MCAF), the monocyte chemoattractant protein-1 gene. A promoter polymorphism in this gene (rs1024611) has earlier been associated with antipsychotic resistance.20 SNP rs4586 is in LD with rs1024611 (D′=1.00, r2=0.62); therefore, it is possible that the association detected could be driven by rs1024611, which is not directly genotyped in this study.
Top hits for PANSS negative, positive and general psychosis
No PANSS change hits were significant after correction for multiple testing. All of the strongest hits were in glutamate receptor genes. The overall top hit was rs7628369 (GRM7 SNP, see above) associating with a change in PANSS negative score (P=0.0002), and the next strongest was the same SNP associating with PANSS general psychosis score. After this, the strongest association was with rs2106190, a GRM8 SNP that was associated with change in positive PANSS score at P=0.0008. The next three strongest hits were all associated with change in PANSS negative score: rs2237547, in GRM3, P=0.0009; rs10512287, in GRIN3A, P=0.0010 and rs757656, in GRM3, P=0.0010. All other associations were above P=0.001. All PANSS change associations below 0.05, along with their locations and associated SNPs of interest, are shown in Supplementary Table S2.
Discontinuation
Study-wide significant association
An SNP in intron 2 of RIMS1, rs502046, was associated with discontinuation from quetiapine due to inefficacy (raw P-value=0.00009; study-wide corrected P=0.021, n=129, odds ratio=3.1; Figure 2). The SNP is in strong LD with a number of other SNPs in introns 2, 3 and 4 (Figure 2). The patients, both European- and African-Americans, carrying the rare variant were more likely to continue with quetiapine treatment than those without (TT: 33/38 discontinued; CT: 36/62 discontinued; CC 13/29 discontinued). To confirm that the association was not caused by stratification beyond that accounted for by the first PCA axis, we repeated the analysis with 10 significant axes of ancestry included as covariates. Again, the resulting raw P-value was still strongly significant at P=0.00027, but when corrected by permutation for all SNPs, this becomes 0.31. This SNP was not associated with discontinuation of any of the other drugs.

Association of RIMS1 SNP rs502046 with quetiapine discontinuation (n=129; uncorrected P=0.00009). The patients carrying the rarer variant were much less likely to discontinue than those without.
Top hits
All significant hits (uncorrected P<0.05), along with their genic position, are shown in Supplementary Table S3. None of the strongest hits for discontinuation from a drug due to inefficacy made any strong biological sense. The top hit for discontinuation from olanzapine (n=141) was an intronic SNP in GRM7 that was associated with various other intronic SNPs within the gene, none of which had any obvious function (proximity to an exon, association with gene expression, location in known regulatory region). The top hit for discontinuation from perphenazine (n=106), the single typical antipsychotic tested in CATIE, was rs2756271, located 1.5 kb upstream of the start of the prion protein gene PRNP, and again the SNP showed no obvious function, nor was it associated with any SNPs closer to the gene. The top hit for discontinuation from risperidone (n=136) was rs10495449, an SNP just downstream from the muscarinic acetylcholine receptor gene CHRM3. The SNP shows no association with any other SNP within the gene and has no obvious function. Finally, the top hit for ziprasidone (n=60) was rs7614915, a synonymous coding SNP in exon 8 of the GRM7 metabotropic glutamate receptor gene.
Weight gain
No weight-gain association stood up to correction for multiple testing. All P-values described are uncorrected.
Top hits
The strongest association with maximum weight gain during phase I was an intronic SNP, rs7164043, in the α7-nicotinic acetylcholine receptor subunit gene CHRNA7 that was associated with quantitative weight gain P=0.0001. The SNP was also associated with change in PANSS total (P=0.006), positive (P=0.01), negative (P=0.03) and general (0.04) scores, change in visuospatial working memory (P=0.01) and change in verbal memory score (0.03). This SNP, however, is only present in African-American populations, with the exception of one patient self-described as ‘White alone’ and identified as of European ancestry by Eigenstrat analysis. Restricting the association of weight to African-Americans increases the significance of the association, such that those carrying the rare variant (n=20) gained an average of 7.02% of their baseline weight, whereas those without the variant allele (n=176) gained, on average, only 0.17% of their baseline weight.
The strongest hit for the weight-gain case–control association test was an intronic SNP in the serotonin receptor gene HTR2A, rs1928042 (P=0.001). This SNP was not associated with the quantitative measure of weight gain, so it is possible that the effect is a false positive. All significant hits (uncorrected P<0.05) for weight gain are displayed in Supplementary Table S4, and the top hits plus others that are of interest due to their genic position are displayed in Table 6.
Genotype imputation analysis
Using tagging SNPs, we found strongly suggestive associations with different SNPs in the GRM7 and GRM8 genes with neurocognitive, PANSS and discontinuation phenotypes, and a strongly suggestive association of an RIMS1 SNP with quetiapine discontinuation. As not all common variants in these genes have been genotyped, it is possible that there are ungenotyped SNPs that are more strongly associated with these phenotypes. We therefore imputed genotypes for all the HapMap SNPs for GRM7, GRM8 and RIMS1 using MaCH (Supplementary Methods). We first checked to see if there was a stronger association with discontinuation from quetiapine for any RIMS1 SNP. We found that the genotyped SNP, rs502046, was the lowest associated SNP in this gene, even after examining associations with all permuted genotypes.
As GRM7 and GRM8 were associated particularly strongly with many phenotypes, we looked to see if imputed SNPs had stronger associations with any phenotype. For most phenotypes, the lowest P-value for an imputed SNP was of the same order of magnitude as that of the most strongly associated genotyped SNP. However, some phenotypes showed much stronger association with an imputed SNP; these are documented in Supplementary Table S1. None of the strongest associated phenotype detailed for GRM7 and GRM8 (above) improved when imputed SNPs were examined.
Previous findings
Where possible, we also checked to see if we were able to replicate other findings of SNPs determining antipsychotic response in schizophrenia. In many cases, this was not possible, as previously associated polymorphisms have been non-SNP variants, such as VNTRs or in/dels. In addition, some important SNPs failed genotyping quality control (see Supplementary Information). Previously associated SNPs that we successfully genotyped here include COMT val108/158met (rs4680), DRD2 -241-A/G (rs1799978), DRD3 Ser9Gly (rs6280) and HTR2A 102-T/C(rs6313). The COMT SNP has been associated with antipsychotic-related improvement in working memory and negative symptoms21, 22 and better treatment response23 (as well as other phenotypes not captured here). We found no significant associations of COMT val108/158met with any phenotype evaluated in this study. The DRD2 SNP −241-A/G has been associated of a faster response to antipsychotic treatment,24 but again we saw no association with any treatment response measures at the time points measured (PANSS scores: 12 weeks; neurocognitive change: 8 weeks). The DRD3 Ser9Gly SNP has earlier been associated with a better response to risperidone treatment in Chinese (carriers of ser allele better response25), worse response to atypical antipsychotic treatment in Caucasians (ser/ser worse than other genotypes26) and improvement in positive symptoms while on olanzapine.27 In this study, the SNP was associated with change in verbal memory at 8 weeks (uncorrected P=0.008), change in visuospatial memory at 8 weeks (uncorrected P=0.03) and discontinuation from risperidone due to inefficacy (uncorrected P=0.02, gly/gly less likely to discontinue due to inefficacy). Finally, the HTR2A 102-T/C SNP has been associated with better response to risperidone in Chinese (CC better response28) and with non-response to antipsychotic treatment in Caucasians (CC overrepresented in non-responders29). In this study, we saw only a very borderline association with change in verbal fluency (uncorrected P=0.05). In summary, although previously associated SNPs and related phenotypes were tested here, we could not provide strong support for the previous associations.
Discussion
This study has identified suggestive associations that if confirmed could be of considerable clinical significance. Notable among these findings is a polymorphism that is strongly associated with discontinuation of quetiapine (rs502046, in RIMS1). When randomized onto quetiapine in phase 1 of the CATIE trial, a minority of patients stayed on the drug (35%) but most discontinued. We have shown that those patients who continued on the drug, presumably responding well, carried a rare variant at the associated locus, and for those patients homozygous for the rare allele, the continuation rate was 55%. If this association is confirmed, quetiapine would likely emerge as a first-line choice for the 22% of patients who carry the favorable genotype. Attempted replication therefore of this association is a priority.
The second study-wide significant association was between a GRM8 variant (rs7778604) and improvement in verbal memory over 8 weeks of antipsychotic treatment. No measure of cognitive improvement, including verbal memory, differed between the medications used in this trial,30 nor did the genetic association with rs7778604 differ significantly between treatment groups. This variant, therefore, appears to convey a general increased responsiveness to the cognitive benefits of antipsychotic treatment. An improved understanding of the mechanisms of this effect could be useful in developing adjunctive medications for improving cognitive response to antipsychotic drugs in schizophrenia. This may have great relevance to the study of cognitive deficits in schizophrenia. Numerous pharmaceutical industry drug-development programs and government efforts, such as the NIH-sponsored project Measurement and Treatment Research to Improve Cognition in Schizophrenia,31 are currently devoted to finding pharmacologic compounds or behavioral strategies that may improve cognitive impairment in schizophrenia.
On balance, these results, like most findings in psychiatric genetics, remain equivocal. There was no single association that stood up to correction for all SNPs and all phenotypes analyzed. However, given the study size and the number of pharmacogenetic questions addressed, there is clearly potential for type II error. This could have been reduced in part by matching particular candidate genes up to the phenotypes that they could most plausibly affect, rather than testing all genes against all phenotypes. However, with the current state of knowledge, we felt that such matching would have to be based at least in part on guesswork and consequently opted for the broader approach presented here. For these reasons, we felt that it was important to make available all associations with uncorrected P-values <0.05, for potential targeted replication by other interested researchers. It should also be noted that as this study was limited to common SNPs there could be unrepresented polymorphisms in these genes with stronger effects on the pharmacogenetic phenotypes than we have found here such as rare SNPs, large structural variants recently implicated in schizophrenia,32, 33 in-dels or VNTRs.
In addition, it is important to appreciate that the CATIE study was not designed with genetic hypotheses in mind, and it is not well powered to test many of the most pressing pharmacogenetic questions. Clinical studies such as CATIE could be an invaluable resource for conducting genetic studies that have direct relevant to patient populations. However, for these studies to be effective, it is an absolute requirement that the genetic component is considered a priori and a set of genetic hypotheses established in advance to ensure sufficient power to explore the relevant questions and reduce the potential for data mining. All study patients should be encouraged to take part in the genetic analyses both to maximize power for the genetic hypotheses and so that the genetic component accurately reflects the overall sample composition. In addition, it is important that psychiatric practitioners facilitate complementary collections in the regular care setting that allow the same hypotheses to be tested and replication analyses to be performed. This is the only way to realize the potential of these trials for advancing personalized medicine.
By contemporary standards for association genetics, nothing in psychiatric genetics has yet qualified as a definitive association.34 A key question for the field is what to do with its results that do not pass contemporary standards. Although we agree that suggestive results need to be widely available, it is difficult to see any advantage in the continued publication of only slightly suggestive results individually, polymorphism by polymorphism and phenotype by phenotype. In many cases, the quality of a gene as a candidate for the condition has made up for very modest P-values to motivate separate publication.14, 35, 36, 37, 38, 39 Large-scale genotyping being carried out here and elsewhere, however, makes clear such an approach is untenable. In our study, there are at least 856 and 368 genetic associations with raw P-values superior to the 0.049 and 0.015 reported for RGS4 SNPs rs2661319 and rs2842030 published in a recent paper35 (see Supplementary Information Tables, noting that the figures 856 and 368 for nominal genetic associations include only the strongest associated SNP per gene to prevent inflation of estimates by LD). Each of these is in genes that can be argued to be plausibly involved (as they were selected as candidate genes). Clearly, it would be both unnecessary and misleading to publish these associations one by one in individual research papers. These considerations demonstrate that the earlier practice in psychiatric genetics of individual reports for suggestive candidate genes is no longer appropriate given the scale of genetic data being generated, including for large numbers of suggestive candidate genes.
The alternative we propose is to report all associations resulting from large-scale screens that both explicitly and implicitly convey the magnitude of testing actually taking place in the community and allow ready interpretation and comparison of the results. To facilitate this effort, we have included as Supplementary Information all raw P-values (<0.05) in a file format that contains the phenotype, SNP and P-value. These results can therefore be easily interrogated by any software tool for comparison with other data sets. With the increase in larger scale genetic studies, and in particular, the increasing prevalence of whole-genome analyses, the risk of producing type II errors when correcting for thousands of SNPs increases exponentially. Many SNPs with real effects will not stand up to correction for study-wide significance. This mandates the use of software programs such as WGAviewer19 to allow the examination and interpretation of results in their genomic context, and necessitates data sharing between studies so that replications of SNPs that may not come out in the top P-values can be detected and followed up.
References
Lieberman JA, Stroup TS, McEvoy JP et al: Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005; 353: 1209–1223.
Bakker PR, van Harten PN, van Os J : Antipsychotic-induced tardive dyskinesia and polymorphic variations in COMT, DRD2, CYP1A2 and MnSOD genes: a meta-analysis of pharmacogenetic interactions. Mol Psychiatry 2008; 13: 544–556.
Reynolds GP : The impact of pharmacogenetics on the development and use of antipsychotic drugs. Drug Discov Today 2007; 12: 953–959.
Bakker PR, van Harten PN, van Os J : Antipsychotic-induced tardive dyskinesia and the Ser9Gly polymorphism in the DRD3 gene: a meta analysis. Schizophr Res 2006; 83: 185–192.
Arranz MJ, Kapur S : Pharmacogenetics in psychiatry: are we ready for widespread clinical use? Schizophr Bull 2008; 34: 1130–1144.
De Luca V, Mueller DJ, de Bartolomeis A, Kennedy JL : Association of the HTR2C gene and antipsychotic induced weight gain: a meta-analysis. Int J Neuropsychopharmacol 2007; 10: 697–704.
Athanasiou MC, Malhotra AK, Xu C, Stephens JC : Discovery and utilization of haplotypes for pharmacogenetic studies of psychotropic drug response. Psychiatr Genet 2002; 12: 89–96.
Arranz MJ, Munro J, Birkett J et al: Pharmacogenetic prediction of clozapine response. Lancet 2000; 355: 1615–1616.
de Leon J, Armstrong SC, Cozza KL : Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47: 75–85.
Grossman I, Sullivan PF, Walley N et al: Genetic determinants of variable metabolism have little impact on the clinical use of leading antipsychotics in the CATIE study. Genet Med 2008; 10: 720–729.
Grossman I : Routine pharmacogenetic testing in clinical practice: dream or reality? Pharmacogenomics 2007; 8: 1449–1459.
Green MF, Kern RS, Heaton RK : Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res 2004; 72: 41.
Braff DL, Greenwood TA, Swerdlow NR, Light GA, Schork NJ : Advances in endophenotyping schizophrenia. World Psychiatry 2008; 7: 11–18.
Sullivan PF, Keefe RS, Lange LA et al: NCAM1 and neurocognition in schizophrenia. Biol Psychiatry 2007; 61: 902–910.
Mohler EG, Shacham S, Noiman S et al: VRX-03011, a novel 5-HT4 agonist, enhances memory and hippocampal acetylcholine efflux. Neuropharmacology 2007; 53: 563–573.
Micale V, Leggio GM, Mazzola C, Drago F : Cognitive effects of SL65.0155, a serotonin 5-HT4 receptor partial agonist, in animal models of amnesia. Brain Res 2006; 1121: 207–215.
Manuel-Apolinar L, Rocha L, Pascoe D, Castillo E, Castillo C, Meneses A : Modifications of 5-HT4 receptor expression in rat brain during memory consolidation. Brain Res 2005; 1042: 73–81.
Lohoff FW, Dahl JP, Ferraro TN et al: Variations in the vesicular monoamine transporter 1 gene (VMAT1/SLC18A1) are associated with bipolar i disorder. Neuropsychopharmacology 2006; 31: 2739–2747.
Ge D, Zhang K, Need AC et al: WGAViewer: software for genomic annotation of whole genome association studies. Genome Res 2008; 18: 640–643.
Mundo E, Altamura AC, Vismara S et al: MCP-1 gene (SCYA2) and schizophrenia: a case–control association study. Am J Med Genet B Neuropsychiatr Genet 2005; 132: 1–4.
Weickert TW, Goldberg TE, Mishara A et al: Catechol-O-methyltransferase val108/158met genotype predicts working memory response to antipsychotic medications. Biol Psychiatry 2004; 56: 677–682.
Bertolino A, Caforio G, Blasi G et al: Interaction of COMT Val108/158 Met genotype and olanzapine treatment on prefrontal cortical function in patients with schizophrenia. Am J Psychiatry 2004; 161: 1798–1805.
Anttila S, Illi A, Kampman O, Mattila KM, Lehtimaki T, Leinonen E : Interaction between NOTCH4 and catechol-O-methyltransferase genotypes in schizophrenia patients with poor response to typical neuroleptics. Pharmacogenetics 2004; 14: 303–307.
Schafer M, Rujescu D, Giegling I et al: Association of short-term response to haloperidol treatment with a polymorphism in the dopamine D(2) receptor gene. Am J Psychiatry 2001; 158: 802–804.
Reynolds GP, Yao Z, Zhang X, Sun J, Zhang Z : Pharmacogenetics of treatment in first-episode schizophrenia: D3 and 5-HT2C receptor polymorphisms separately associate with positive and negative symptom response. Eur Neuropsychopharmacol 2005; 15: 143–151.
Szekeres G, Keri S, Juhasz A et al: Role of dopamine D3 receptor (DRD3) and dopamine transporter (DAT) polymorphism in cognitive dysfunctions and therapeutic response to atypical antipsychotics in patients with schizophrenia. Am J Med Genet B Neuropsychiatr Genet 2004; 124: 1–5.
Staddon S, Arranz MJ, Mancama D, Mata I, Kerwin RW : Clinical applications of pharmacogenetics in psychiatry. Psychopharmacology (Berl) 2002; 162: 18–23.
Lane HY, Chang YC, Chiu CC, Chen ML, Hsieh MH, Chang WH : Association of risperidone treatment response with a polymorphism in the 5-HT(2A) receptor gene. Am J Psychiatry 2002; 159: 1593–1595.
Joober R, Benkelfat C, Brisebois K et al: T102C polymorphism in the 5HT2A gene and schizophrenia: relation to phenotype and drug response variability. J Psychiatry Neurosci 1999; 24: 141–146.
Keefe RS, Bilder RM, Davis SM et al: Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE trial. Arch Gen Psychiatry 2007; 64: 633–647.
Marder SR, Fenton W : Measurement and treatment research to improve cognition in schizophrenia: NIMH MATRICS initiative to support the development of agents for improving cognition in schizophrenia. Schizophr Res 2004; 72: 5–9.
Stone JL, O’Donovan MC, Gurling H et al: Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008; 455 (7210): 237–241.
Walsh T, McClellan JM, McCarthy SE et al: Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008; 320: 539–543.
McCarthy MI, Abecasis GR, Cardon LR et al: Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008; 9: 356–369.
Campbell DB, Ebert PJ, Skelly T et al: Ethnic stratification of the association of RGS4 variants with antipsychotic treatment response in schizophrenia. Biol Psychiatry 2008; 63: 32–41.
Crowley JJ, Keefe RS, Perkins DO, Stroup TS, Lieberman JA, Sullivan PF : The neuregulin 1 promoter polymorphism rs6994992 is not associated with chronic schizophrenia or neurocognition. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 1298–1300.
Campbell DB, Lange LA, Skelly T, Lieberman J, Levitt P, Sullivan PF : Association of RGS2 and RGS5 variants with schizophrenia symptom severity. Schizophr Res 2008; 101: 67–75.
Pinheiro AP, Keefe RS, Skelly T et al: AKT1 and neurocognition in schizophrenia. Aust N Z J Psychiatry 2007; 41: 169–177.
Sullivan PF, Keefe RS, Lange LA et al: NCAM1 and neurocognition in schizophrenia. Biol Psychiatry 2007; 61: 902–910.
Acknowledgements
We thank Kevin Shianna and team for genotype data, Jeff Dawson for management of the phenotype database and Mike Weale for tagging. We are indebted to the patients and families who took part in this study. We also thank Sonia Davies for her help in providing and interpreting the CATIE phenotype data. The CATIE project was supported by NIMH contract N01 MH90001.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Need, A., Keefe, R., Ge, D. et al. Pharmacogenetics of antipsychotic response in the CATIE trial: a candidate gene analysis. Eur J Hum Genet 17, 946–957 (2009). https://doi.org/10.1038/ejhg.2008.264
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.264
Keywords
This article is cited by
-
Ethnic Differences in Antipsychotic Response: What Genetic Variation Does and Does Not Tell Us
Current Behavioral Neuroscience Reports (2023)
-
Gene expression and response prediction to amisulpride in the OPTiMiSE first episode psychoses
Neuropsychopharmacology (2020)
-
Variants of GRM7 as risk factor and response to antipsychotic therapy in schizophrenia
Translational Psychiatry (2020)
-
A functional HTR1A polymorphism, rs6295, predicts short-term response to lurasidone: confirmation with meta-analysis of other antipsychotic drugs
The Pharmacogenomics Journal (2020)
-
Pharmacogenetic Correlates of Antipsychotic-Induced Weight Gain in the Chinese Population
Neuroscience Bulletin (2019)
