
Abstract
In a study of 530 individuals with non-syndromic, sensorineural hearing loss, we identified 18 mutations at connexin 26 (Cx26), four of which are novel (−23G>T, I33T, 377_383dupTCCGCAT, W172R) and the remaining 14 (ivs1+1G>A, M1V, 35delG, W24X, I35S, V37I, R75W, W77X, 312del14, E120del, Q124X, Y136X, R143W, R184P) being mutations previously described. To gain insight into functional consequences of these mutations, cellular localization of the mutant proteins and their ability to permit lucifer yellow transfer between cells was studied in seven of them (W24X, I33T, I35S, R75W, E120del, W172R and R184P). I35S and R184P showed impaired trafficking of the protein to the plasma membrane. I33T, R75W, E120del and W172R showed predominantly membrane localization but did not form functional gap junction channels. Surprisingly, W24X, a protein-truncating mutation, apparently permits formation of a full-length protein, perhaps due to a stop codon read-through mechanism. These results provide further evidence that Cx26 mutations affect gap junction activity by mis-regulation at multiple levels.
Similar content being viewed by others

Introduction
Hereditary hearing loss affects about 1 in 2000 newborns.1 Genetic causes of hearing loss are quite heterogeneous and to date 42 genes associated with non-syndromic hearing loss have been identified (http://webhost.ua.ac.be/hhh/). Among them, connexin 26 (Cx26 or GJB2) is the major known cause of hearing loss in Europe,2, 3, 4 the United States5 and Japan.6 Mutations at Cx26 are associated with both recessive7 and dominant8 forms of non-syndromic hearing loss. 35delG, 167delT and 235delC are the most common Cx26 mutations in southern European,4 Ashkenazi Jewish9, 10 and Far-Eastern6 populations, respectively. Cx26 encodes a gap junction protein known to form intercellular channels by the juxtaposition of connexon subunits between two adjacent cells. In the inner ear, these channels are expressed in the supporting cells, spiral limbus, stria vascularis and spiral ligament11 and permit transport of fluids and small molecules of less than 1 kDa.12 It has been proposed that potassium ions needed for the initiation of action potentials in the hair cells and inositol 1,4,5-trisphosphate, a Ca2+-mobilizing messenger are transported through certain gap junction channels of the supporting cells and stria vascularis.13, 14, 15 The three-dimensional structure of M34A, a human Cx26 mutation, reveals a prominent electron density in the pore of each hemichannel, suggesting that physical blocking may be important in gap junction channel regulation.16
Although W24X is known to be a common Cx26 mutation in India,17, 18 the full spectrum of mutations of this gene occurring in India is not known. We present here the results of a study of Cx26 in 530 individuals exhibiting non-syndromic, sensorineural hearing loss, in which we identified four novel Cx26 mutations and 14 mutations that have been described earlier. For a subset of the mutations identified, we also examined their effect on steady state protein levels, cellular localization of the mutant proteins and their ability to form functional gap junctions in a cell-based, lucifer yellow dye transfer assay.
Methods
Subjects
Five hundred and thirty probands exhibiting severe-to-profound hearing loss were ascertained. They belonged to apparently unrelated families and had at least one additional sibling manifesting hearing loss. A complete clinical history of each proband was taken to ensure that the hearing loss was not a result of infection, acoustic trauma, ototoxic drugs or premature birth. Hearing levels of subjects were assessed by pure tone audiometry including bone conduction tests using a calibrated audiometer. Hearing thresholds were obtained at 250 Hz, 500 Hz, 1000 Hz, 2000 Hz, 4000 Hz and 8000 Hz in a sound-treated room. A 10 millilitre blood sample was collected from affected and unaffected individuals of the families after obtaining written informed consent. The control samples used in the study were from individuals with normal hearing from the southern, northern and western parts of India and these individuals did not have a family history of hearing loss. This work had the approval of the institutional bioethics and biosafety committees of the participating centres.
Mutation analysis
The coding exon (exon 2) of Cx26, was amplified from genomic DNA with two partially overlapping sets of primers: Cx26–1F (5′-TCT TTT CCA GAG CAA ACC GC-3′) and Cx26–1R (5′-GAC ACG AAG ATC AGC TGC AG-3′); Cx26–2F (5′-CCA GGC TGC AAG AAC GTG TG-3′) and Cx26–2R (5′-GGG CAA TGC GTT AAA CTG GC-3′), which amplify DNA fragments of 286 bp and 519 bp respectively. Amplification of the non-coding exon (exon 1) and the flanking donor splice site sequences were performed using Advantage-GC genomic PCR kit (Clontech) and PCR primers Cx26–3F (5′-TCC GTA ACT TTC CCA GTC TCC GAG GGA AGA GG-3′) and Cx26–3R (5′-CCC AAG GAC GTG TGT TGG TCC AGC CCC-3′). The PCR products were purified using Qiaquick columns (Qiagen) and were directly sequenced using ABI PRISM BigDye Terminator Cycle Sequencing kit v3.1 on an ABI3100 Genetic Analyzer (Applied Biosystems).
Molecular cloning
The coding region of Cx26 was PCR-amplified using primer pairs Nhe1-Cx26-F (5′-AAC CGC GCT AGC GCC ACC ATG GAT TGG GGC ACG CTG CA-3′) and Xho1-Cx26-R (5′-GCC GCA CTC GAG ATT GGG CAA TGC GTT AAA CT-3′) from the genomic DNA of individuals carrying specific mutant alleles. The amplified products were cloned in the pIRES2-EGFP (Clontech) vector using the Nhe1 and Xho1 restriction enzyme sites. pIRES2-EGFP contains an internal ribosome entry site (IRES) located between the multiple cloning site and the enhanced green fluorescent protein (EGFP) coding region, permitting Cx26 and EGFP proteins to be made from a single bicistronic mRNA and, hence, allowing for efficient selection of transfected cells for functional studies. For cloning in pEGFP-N1 (Clontech), PCR amplification of the coding region of the Cx26 alleles was performed using the primers Nhe1-Cx26-F and Xho1-Cx26-FUS-R (5′-GGA TCC CTC GAG AAC TGG CTT TTT TGA CTT CCC-3′). In this case, the Cx26 alleles were cloned in frame with the EGFP coding sequences using the Nhe1 and Xho1 restriction enzyme sites to obtain fusions to the N Terminus of EGFP. All constructs used in this study were confirmed by sequencing to ensure that no additional sequence alterations were incorporated during PCR.
Cell culture and transfection
A communication-deficient HeLa cell line, which has a very low endogenous level of intercellular dye transfer activity (provided by Professor Klaus Willecke, University of Bonn, Germany) was cultured. For transfection, cells were seeded in 35 mm dishes containing 12 mm polylysine-coated cover slips (for immunocytochemistry and intercellular dye transfer assays) or without cover slips (for protein immunoblot analysis). All transfections were performed using Lipofectamine 2000 (Invitrogen), and experiments were done 36 h after transfection.
Protein immunoblot analysis
Cell lysate was obtained by treating cells with 200 μl of lysis buffer (60 mM Tris pH 6.8, 3% SDS with protease inhibitor cocktail). The cell lysate was passed through a syringe with a 28-G needle and incubated at 4°C for 30 min. The lysate was then spun and the supernatant stored at −80°C till further experiments. Here, 5 μl of the supernatant was used for determination of protein concentration using the bicinchoninic acid method (Sigma). Then 100 μg of protein was precipitated using trichloroacetic acid. The pellet was washed with ice cold acetone, resuspended in 1X SDS loading buffer, heated to 100°C for 5 min and separated in a denaturing 12% polyacrylamide gel, along with pre-stained protein markers (New England Biolabs), followed by electro-blotting to a nitrocellulose membrane. Blocking of the membrane was preformed with 3% BSA in PBS at 4°C for about 14 h. A polyclonal antibody against cytoplasmic loop of rat connexin 26 (Zymed) was used as the primary antibody at a dilution of 1:500. Primary and secondary antibody incubations were done for 3 h each at 4°C in PBS containing 1% BSA and 0.05% Tween 20. Washes were done with PBS containing 0.05% Tween 20 at room temperature. Blots were developed using the enhanced chemiluminescence system (Pierce). For western blots to detect GFP, polyclonal antibodies against GFP (Affinity BioReagents) were used at a dilution of 1:2000.
Immunocytochemistry
The cells were washed with PBS, fixed with absolute ethanol for 10 min, washed again with PBS and incubated with blocking solution (5% normal goat serum, 0.1%. Triton X-100 in PBS) for 1 h at room temperature. Polyclonal antibody against rat connexin 26 (Zymed) was diluted (1:100) in blocking solution and incubation was carried out for 1 h at room temperature. The cells were washed with blocking solution and incubated for 45 min with a 1:400 dilution of a secondary antibody conjugated with Alexa 568 (Molecular Probes). The cells were then washed with PBS, mounted on a glass slide using an aqueous mounting medium (Sigma) and imaged using a Carl Zeiss LSM 510 Meta confocal laser scanning microscope with the × 63/1.4 oil immersion objective.
Intercellular dye transfer
After a fresh culture medium change, lucifer yellow (MW: 457 Da, Sigma) was microinjected into cells as a 5% (wt/vol) solution in 0.33 M LiCl. Microinjections were administered in a single cell located in clusters of cells expressing GFP, using FemtoJet and InjectMan NI2 microinjection system (Eppendorf). Forty or more microinjections were administered for each mutation analysed. Imaging was performed before injection and 5 min after the injection, using a Leica DM IRB epifluorescent microscope with the × 40/0.60 objective. Non-transfected HeLa cells on the same cover slip were injected as controls.
Results
Cx26 mutations
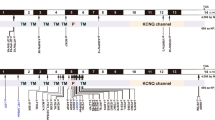
The entire coding region of Cx26 was sequenced in the 530 individuals manifesting severe-to-profound hearing loss. For those individuals who were found to be heterozygous for any mutation in the coding region of the gene, the non-coding exon of Cx26 was sequenced and examined. Four novel (−23G>T, I33T, 377–383dupTCCGCAT, W172R) and 14 previously known (ivs1+1G>A, M1V, 35delG, W24X, I35S, V37I, R75W, W77X, 312del14, E120del, Q124X, Y136X, R143W, R184P) Cx26 mutations were detected (Table 1A). Sixteen Cx26 mutations lie in the coding exon, one in the non-coding exon and one in the intron. Of the 16 mutations in the coding exon, eight were missense mutations, four were nonsense mutations and four deletions or duplications of 1–14 nucleotides. Cx26 mutations were observed in 24% (128/530) of the individuals exhibiting hearing loss. About 21% (112/530) of the individuals studied harboured bi-allelic Cx26 mutations. W24X was the most common mutation in this group of individuals, with an allele frequency of 0.164 (16.4%). Of the four novel mutations identified, two were missense mutations (I33T, W172R), one mutation involved a 7 bp duplication (377_383 dupTCCGCAT) and the fourth one (−23G>T) was located in exon 1. The exon 1 mutation (−23G>T) was observed in a compound heterozygous state along with W24X. The nucleotide, −23G lies at the −1 position with respect to the splice donor site of the intron 1 of Cx26. Using the splice site prediction by NatGene2 (http://www.cbs.dtu.dk/services/NetGene2/),19, 20 −23G>T is predicted to affect the regulation of the splice donor site usage and the actual splice site may be lost due to this change. Two individuals were heterozygous for R75W, which was previously reported to be associated with dominant hearing loss and palmoplantar keratoderma.21 Richard et al21 found R75W in a heterozygous condition in an individual from the control group without skin disorder. Hearing levels of the control group individuals in this study were unknown, and individuals affected with hearing loss were likely to be present in the control group they studied. In our study, the two individuals who were heterozygous for R75W exhibited non-syndromic hearing loss, but no apparent skin abnormality. We also found six known single nucleotide polymorphisms (V27I, E114G, R127H, V153I, D159Y, R165W) and a novel single nucleotide polymorphism (F106F) (Table 1B). A novel and rare sequence variant, M195I was observed in a heterozygous condition in one proband; its pathogenic status remains uncertain (Table 1C). As both methionine and isoleucine are nonpolar neutral amino acid residues, M195I may be a non-pathogenic variant.
Expression of the mutant connexin 26 proteins
We estimated steady state protein levels by western analysis using total cell lysates for the 7 Cx26 mutations identified, W24X, I33T, I35S, R75W, E120del, W172R, R184P (Figure 1a). A very low endogenous expression of the Cx26 protein was observed in HeLa cells transfected with the empty vector and steady state protein level was highest in the case of HeLa cells transfected with wild-type Cx26 allele. The seven Cx26 mutant alleles yielded full-length proteins of the expected 26 kDa size. For W24X, which is known to create a premature stop codon at the 24th amino acid, detection of a full-length protein of 26 kDa was unexpected. We therefore analysed it further by testing the possibility of a stop codon read-through mechanism for the formation of full-length protein. This was done by analysing the formation of EGFP in a construct with the W24X carrying sequence fused to the N terminus of EGFP. Characteristic membrane localization of EGFP was observed for the wild-type Cx26-EGFP fusion protein. However, for the W24X-EGFP fusion, EGFP exhibited cytoplasmic localization (Figure 1b). As W24X mutation is located upstream of EGFP, the expression of EGFP may be due to a mechanism involving stop codon read-through. To test the possibility of EGFP being translated from its own start codon in case of WT-EGFP and W24X-EGFP constructs, western blot analysis with anti-GFP antibody was carried out (Figure 1c). GFP is known to be a 27 kDa protein and the Cx26-GFP fusion protein has an apparent molecular weight of about 53 kDa. We observed a band corresponding to 27 kDa in case of transfection with pEGFP-N1 and not with WT-EGFP or W24X-EGFP. This suggests that EGFP is apparently not made from its own start codon in case of WT-EGFP and W24X-EGFP.

(a) Western analysis of expression of the normal and seven mutant Cx26 proteins in communication-deficient HeLa cells; (b) HeLa cells transfected with pEGFP-N1 empty vector (left), fusion construct of normal Cx26 with EGFP (middle) and fusion construct of the W24X allele of Cx26 with EGFP (right) and (c) western analysis of HeLa cells transfected with empty vector, WT-EGFP and W24X-EGFP.
Cellular localization of the mutant Cx26 proteins
In immunocytochemistry experiments aimed at examining cellular localization of the mutant Cx26 proteins (Figure 2), the wild-type Cx26 exhibited characteristic punctate staining in the plasma membrane. On the other hand, W24X exhibited a predominantly cytoplasmic localization. Tryptophan at codon 24 lies in the first transmembrane domain whereas the anti-connexin antibody used is known to recognize the cytoplasmic loop between second and third transmembrane domain. So, even though W24X apparently makes a full-length protein in the cells used, its cellular localization is not normal. I35S and R184P also exhibit defective localization, as the mutant proteins were found largely in the cytoplasm. I33T, R75W and W172R exhibited membrane localization similar to wild-type Cx26. E120del mutant exhibited a predominantly membrane localization, but a small amount of protein was observed in the cytoplasm as well.

Cellular localization of the normal and mutant Cx26 proteins in HeLa cells (Scale bar: 10 μm). For each mutation, the image on the left and right correspond to fluorescence and phase contrast, respectively.
Dye transfer assay involving Cx26 mutations
Cx26 mutations (I33T, R75W, E120del and W172R), which exhibited normal or nearly normal trafficking to the plasma membrane were analysed by dye transfer assays using HeLa cell clusters (Figure 3). Although intercellular transfer of lucifer yellow between EGFP-positive cells was observed for wild-type Cx26 allele, no such dye transfer was observed in case of I33T, E120del and W172R mutations. These three mutations are associated with recessive non-syndromic hearing loss. In the case of R75W, which is associated with dominant hearing loss, transfections were done using R75W and wild-type Cx26 DNA in equal molar ratios and also, R75W without the wild-type Cx26 DNA. No dye transfer was observed in both these instances, suggesting a possible dominant-negative effect. So, even though the I33T, E120del, W172R and R75W proteins localized apparently normally to the plasma membrane, gap junction channels formed by these showed defective activity. For the mutant proteins, which exhibited defective cellular localization, dye transfer assays were not carried out.

Lucifer yellow dye transfer analysis. (a) Left panel shows GFP-positive cells before the dye injection and the right panel shows microinjected lucifer yellow, 5 min post injection. Microinjected cells are highlighted with asterisks and (b) number of injections carried out are shown. Positive injections are the number of instances in which Lucifer yellow diffused from the injected cell to at least two neighbouring cells.
Discussion
Cx26 mutations were found in 24% of the 530 individuals with hearing loss that we studied. This frequency is much lower than that observed among hearing impaired individuals in, for example, European and North American populations, where Cx26 mutations account for 30–60% of autosomal recessive forms of hearing loss.13 It has been suggested that the high frequency of Cx26 mutations in European and American populations is due to assortative mating among deaf individuals.22, 23 Marriages between deaf individuals are rare in India. This factor, along with the high prevalence of consanguineous marriages, could be an explanation for the relatively low frequency of Cx26 mutations in India. This possibility is supported by the finding that the frequency of Cx26 mutations among the Pakistani populations is about three-fold lower than that observed in India and consanguinity is about two times more frequent in Pakistan than in India.24 Taken together, these results indicate that at least as far as Cx26 is concerned, assortative mating and consanguinity may have opposing effects with respect to mutation frequency. Further epidemiological studies on Cx26 mutations should throw more light on this issue. The results of this study show that the spectrum of Cx26 mutations observed in India is different from that in the European, Ashkenazi Jewish and Far-Eastern populations. W24X was the most common mutation observed in this study and it accounted for 73% of all pathological mutations at this gene. The W24X mutation has a high carrier frequency of 0.024 (2.4%) and is a founder mutation in the Indian population.17 A question that arises here is whether the high carrier frequency of the W24X mutation is associated with a heterozygote advantage. This seems to be the case for R143W, which is associated with recessive non-syndromic hearing loss. It appears that R143W is associated with a skin phenotype that might counter balance an evolutionary disadvantage caused by hearing loss. R143W heterozygotes have normal hearing and exhibit epidermal thickening. R143W homozygotes have hearing loss and exhibit epidermal thickening and sweat hyperosmolarity.25 This thickening of epidermis is perhaps due to increased cell survival mediated by R143W.26 Future studies could test if W24X also confers a heterozygote advantage. Of the 530 individuals screened, one was a compound heterozygote for W24X and R143W. We also observed that out of the 1060 chromosomes studied, only five chromosomes harboured the 35delG mutation, which is the most common mutation in European populations. 167delT and 235delC, which are the most common mutations in the Ashkenazi Jewish populations and Japanese populations respectively, were not observed in our study. Our results indicate that the R75W mutation is most likely associated with the non-syndromic form of hearing loss rather than the syndromic hearing loss as reported earlier,21 although the effect of genetic background cannot be ruled out. Identification of 18 mutations and seven polymorphisms in this study and a large spectrum of mutations and polymorphisms identified by earlier studies on Cx26, suggest that this gene is subject to a relaxed selection.
Non-syndromic hearing loss is the most heterogeneous genetic disorder known, with over a hundred loci already implicated. Different mutations in the same gene are known to result in dominant or recessive hearing loss. Individuals within the same family harbouring the same mutation may exhibit significant differences in hearing levels.27 Cell biological studies have been the method of choice for understanding Cx26 function.28 These studies have revealed that the various Cx26 mutations associated with hearing loss have distinct assembly, trafficking and gating properties that are different from the wild type.29, 30 The cellular localization and lucifer yellow dye transfer properties of the V84L mutation in Cx26 are indistinguishable from those of the wild type, but this mutant exhibits impaired permeability to the Ca2+-mobilizing messenger inositol 1,4,5-triphosphate [Ins(1,2,5)P3] that results in a reduced ability to propagate Ca2+ waves.14 Our functional studies on Cx26 mutations also showed that the various mutants have distinct cellular phenotypes. Both the isoleucines at codon 33 and codon 35 lie in the first transmembrane domain, which is involved in voltage sensor gating (Figure 4). The I33T mutation exhibits normal membrane localization but defective gap junction channel activity, whereas I35S exhibited predominantly cytoplasmic localization. Both threonine and serine are uncharged polar amino acids, which differ only by a methyl group, but their substitution with two closely spaced isoleucines results in different cellular localization phenotypes. Arginine 75 resides in the second transmembrane domain, which has a role in the oligomerization of connexins to form the hexameric connexon hemichannels. An earlier study using insect Sf9 cells has reported that R75W exhibits oligomerization defects.31 The R75W mutation exhibits normal trafficking to the plasma membrane but has defective gap junction activity. Co-transfection of R75W with wild-type Cx26 DNA did not result in the rescue of the mutant phenotype suggesting the dominant-negative effect of this mutation. The E120del mutation results in the deletion of a glutamic acid from a stretch of two non-conserved glutamic acids in the cytoplasmic loop region. The residues in the intracellular loop region and C-termini are highly variable among different connexins and are hence thought to be responsible for regulation.12 These residues could impart unique properties to the various connexin molecules. Tryptophan 172 and Arginine 184 are located in the second extracellular loop region. Each extracellular domain contains a characteristic arrangement of three cysteine residues that help the two opposing connexons to dock with each other. The R184P mutation results in the substitution of a conserved arginine to a proline, and exhibits defective trafficking to the plasma membrane. The W172R mutation results in the substitution of a non-conserved tryptophan to an arginine and exhibits normal plasma membrane localization but defective gap junction channel activity. Tryptophan 172 resides in close proximity to two of the three conserved signature cysteine residues of the extracellular loop. We suggest that the W172R mutation could result in defective docking of the two opposing connexon hemichannels. W24X was not expected to form a protein, but surprisingly we observed an apparently full-length protein, whose cellular localization was, however, defective. It is possible that the stop codon read-through mechanism seen in the HeLa cells is a consequence of the cellular expression system used and does not necessarily reflect the in vivo situation. The in vivo effect of W24X on the Cx26 protein can be tested if cochlear tissue harbouring this mutation is available for study. Our results thus indicate that the Cx26 mutations associated with hearing loss affects gap junction activity by multiple mechanisms but lead to an almost identical clinical phenotype of severe-to-profound hearing loss.

Schematic representation of the Cx26 protein, and mutations identified. The extent of amino acid conservation is colour-coded. Residues shown in shades of blue (1–2) are not conserved and are rapidly evolving. Residues in white (3–6) show an average degree of conservation and residues in shades of red (7–9) are highly conserved and are slowly evolving. The degree of conservation of the polymorphic residues was analysed using ConSeq (http://conseq.tau.ac.il), the sequence only variant of Rate4Site, an algorithmic tool for the identification of functional regions in proteins.32
References
Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE : Genetic epidemiological studies of early-onset deafness in the US school-age population. Am J Med Genet 1993; 46: 486–491.
Denoyelle F, Weil D, Maw MA et al: Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet 1997; 6: 2173–2177.
Zelante L, Gasparini P, Estivill X et al: Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet 1997; 6: 1605–1609.
Estivill X, Fortina P, Surrey S et al: Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998; 351: 394–398.
Kelley PM, Harris DJ, Comer BC et al: Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998; 62: 792–799.
Ohtsuka A, Yuge I, Kimura S et al: GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum Genet 2003; 112: 329–333.
Kelsell DP, Dunlop J, Stevens HP et al: Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997; 387: 80–83.
Denoyelle F, Lina-Granade G, Plauchu H et al: Connexin 26 gene linked to a dominant deafness. Nature 1998; 393: 319–320.
Morell RJ, Kim HJ, Hood LJ et al: Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med 1998; 339: 1500–1505.
Sobe T, Erlich P, Berry A et al: High frequency of the deafness-associated 167delT mutation in the connexin 26 (GJB2) gene in Israeli Ashkenazim. Am J Med Genet 1999; 86: 499–500.
Lautermann J, Frank HG, Jahnke K, Traub O, Winterhager E : Developmental expression patterns of connexin26 and -30 in the rat cochlea. Dev Genet 1999; 25: 306–311.
Kumar NM, Gilula NB : The gap junction communication channel. Cell 1996; 84: 381–388.
Petit C, Levilliers J, Hardelin JP : Molecular genetics of hearing loss. Annu Rev Genet 2001; 35: 589–646.
Beltramello M, Piazza V, Bukauskas FF, Pozzan T, Mammano F : Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat Cell Biol 2005; 7: 63–69.
Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X : Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci USA 2005; 102: 15201–15206.
Oshima A, Tani K, Hiroaki Y, Fujiyoshi Y, Sosinsky GE : Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc Natl Acad Sci USA 2007; 104: 10034–10039.
RamShankar M, Girirajan S, Dagan O et al: Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J Med Genet 2003; 40: e68.
Maheshwari M, Vijaya R, Ghosh M, Shastri S, Kabra M, Menon PS : Screening of families with autosomal recessive non-syndromic hearing impairment (ARNSHI) for mutations in GJB2 gene: Indian scenario. Am J Med Genet 2003; 120: 180–184.
Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S : Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res 1996; 24: 3439–3452.
Brunak S, Engelbrecht J, Knudsen S : Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 1991; 220: 49–65.
Richard G, White TW, Smith LE et al: Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet 1998; 103: 393–399.
Nance WE, Kearsey MJ : Relevance of connexin deafness (DFNB1) to human evolution. Am J Hum Genet 2004; 74: 1081–1087.
Nance WE, Liu XZ, Pandya A : Relation between choice of partner and high frequency of connexin-26 deafness. Lancet 2000; 356: 500–501.
Santos RL, Wajid M, Pham TL et al: Low prevalence of Connexin 26 (GJB2) variants in Pakistani families with autosomal recessive non-syndromic hearing impairment. Clin Genet 2005; 67: 61–68.
Meyer CG, Amedofu GK, Brandner JM, Pohland D, Timmann C, Horstmann RD : Selection for deafness? Nat Med 2002; 8: 1332–1333.
Common JE, Di WL, Davies D, Kelsell DP : Further evidence for heterozygote advantage of GJB2 deafness mutations: a link with cell survival. J Med Genet 2004; 41: 573–575.
Cohn ES, Kelley PM, Fowler TW et al: Clinical studies of families with hearing loss attributable to mutations in the connexin 26 gene (GJB2/DFNB1). Pediatrics 1999; 103: 546–550.
Elfgang C, Eckert R, Lichtenberg-Frate H et al: Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J Cell Biol 1995; 129: 805–817.
Martin PE, Coleman SL, Casalotti SO, Forge A, Evans WH : Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal heriditary deafness. Hum Mol Genet 1999; 8: 2369–2376.
Thonnissen E, Rabionet R, Arbones ML, Estivill X, Willecke K, Ott T : Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum Genet 2002; 111: 190–197.
Oshima A, Doi T, Mitsuoka K, Maeda S, Fujiyoshi Y : Roles of Met-34, Cys-64, and Arg-75 in the assembly of human connexin 26. Implication for key amino acid residues for channel formation and function. J Biol Chem 2003; 278: 1807–1816.
Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N : Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 2002; 18 (Suppl 1): S71–S77.
Acknowledgements
We thank all the subjects who participated in this study. We thank Prof Klaus Willecke (University of Bonn, Germany) for kindly providing communication-deficient HeLa cells and Prof Sharat Chandra for critical comments on the paper. We thank Suma for help with the confocal microscopy, Arunima for DNA sequencing and Rahul for western analysis. Funds for the work were provided by DBT, New Delhi, AYJNIHH, Mumbai and JNCASR, Bangalore. RSM and AG acknowledge research fellowships from CSIR, New Delhi.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
None.
Rights and permissions
About this article
Cite this article
Mani, R., Ganapathy, A., Jalvi, R. et al. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur J Hum Genet 17, 502–509 (2009). https://doi.org/10.1038/ejhg.2008.179
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.179
Keywords
This article is cited by
-
Confirmation of COL4A6 variants in X-linked nonsyndromic hearing loss and its clinical implications
European Journal of Human Genetics (2022)
-
Comparative analysis of functional assay evidence use by ClinGen Variant Curation Expert Panels
Genome Medicine (2019)
-
Elucidation of the unique mutation spectrum of severe hearing loss in a Vietnamese pediatric population
Scientific Reports (2019)
-
Microarray-based screening system identifies temperature-controlled activity of Connexin 26 that is distorted by mutations
Scientific Reports (2019)
-
Connexinopathies: a structural and functional glimpse
BMC Cell Biology (2016)
