
Abstract
Kallmann syndrome is a developmental disease that combines hypogonadotropic hypogonadism and anosmia. Putative loss-of-function mutations in PROKR2 or PROK2, encoding prokineticin receptor-2 (a G protein-coupled receptor), and one of its ligands, prokineticin-2, respectively, have recently been reported in approximately 10% of Kallmann syndrome affected individuals. Notably, given PROKR2 mutations were found in the heterozygous, homozygous, or compound heterozygous state in patients, thus raising the question of a possible digenic inheritance of the disease in heterozygous patients. Indeed, one of these patients was also carrying a missense mutation in KAL1, the gene responsible for the X chromosome-linked form of Kallmann syndrome. Mutations in PROK2, however, have so far been found only in the heterozygous state. Here, we report on the identification of PROK2 biallelic mutations, that is, a missense mutation, p.R73C, and a frameshift mutation, c.163delA, in two out of 273 patients presenting as sporadic cases. We conclude that PROK2 mutations in the homozygous state account for a few cases of Kallmann syndrome. Moreover, since the same R73C mutation was previously reported in the heterozygous state, and because Prok2 knockout mice exhibit an abnormal phenotype only in the homozygous condition, we predict that patients carrying monoallelic mutations in PROK2 have another disease-causing mutation, presumably in still undiscovered Kallmann syndrome genes.
Similar content being viewed by others


Introduction
Kallmann syndrome (KS) combines hypogonadotropic hypogonadism and anosmia or hyposmia, that is, a deficiency of the sense of smell.1 Anosmia/hyposmia is related to the absence or hypoplasia of the olfactory bulbs and tracts.2 Hypogonadism is due to deficiency in gonadotropin-releasing hormone,3 and probably results from a failure of the embryonic migration of gonadotropin-releasing hormone-synthesising neurones to the forebrain.4 There are familial cases, but most patients present as seemingly sporadic cases. This is a genetically heterogeneous disease. Loss-of-function mutations in KAL1 (NCBI GeneID: 3730)5, 6, 7 and FGFR1 (NCBI GeneID: 2260)8 account for the X chromosome-linked form and an autosomal dominant form of the disease, respectively. KAL1 encodes anosmin-1, a locally restricted glycoprotein of embryonic extracellular matrices,9 which is likely to be involved in FGF-signalling through FGFR1.10 Mutations in KAL1 or FGFR1 are found in approximately 20% of all KS patients (see).11
In 2006, we identified point mutations in either PROK2 (NCBI GeneID: 60675) or PROKR2 (NCBI GeneID: 128674), encoding prokineticin-2 and prokineticin receptor-2 (a G protein-coupled receptor), respectively, in a further 10% of KS patients, both familial and sporadic cases.12 A majority of these individuals had only one mutated allele. However, we found four patients who were carrying different PROKR2 mutations in the homozygous state (two patients) or compound heterozygous state (two patients). Notably, the two biallelic mutations were also detected in the heterozygous state in other unrelated patients. The large excess of heterozygous patients for PROKR2 mutations versus homozygotes and compound heterozygotes, combined with the observation that only homozygous knockout mice reproduce the KS phenotype,13 led us to consider the possibility of a digenic or oligogenic mode of transmission of the human disease, especially in heterozygous patients. Although PROK2 was an obvious candidate, we did not find a mutation of this gene in any of the patients carrying a mutated PROKR2 allele. One of these patients, however, was also carrying a missense mutation in KAL1, indicating probable digenic inheritance in this individual.12 Notably, a family has been reported since then, in which a KS male was carrying both a deleterious missense mutation in FGFR1 and an intronic deletion in NELF (NCBI GeneID: 26012) that affected splicing.14
Biallelic mutations in PROK2 have not been reported so far, thus leaving open the question of whether a digenic transmission mode should also be expected in KS individuals carrying only one mutated PROK2 allele. Here, we report on two KS patients presenting as sporadic cases, who carry different PROK2 mutations in the homozygous state. These are a frameshift mutation, and a missense mutation we had previously reported in the heterozygous state.
Patients
Case 1
Patient A is a male of Turkish origin, born of consanguineous parents (see Figure 1). He was referred at the age of 18 because of the absence of spontaneous puberty. In addition, he suffered from anosmia. On clinical examination, he had a micropenis and infantile testes. The olfactory bulbs could not be visualised by MRI. Endocrinological tests confirmed the presence of hypogonadotropic hypogonadism in the patient.

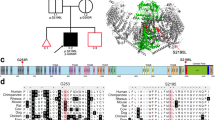
Pedigrees of the two KS patients. Open symbols denote clinically unaffected individuals. Filled symbols denote affected individuals with both hypogonadism and anosmia. The PROK2 genotypes, if available, are indicated below. The symbol + denotes normal allele. Electrophoretograms of the PROK2 mutated sequences are shown. The asterisk indicates the position of the nucleotide deleted in patient B.
Case 2
Patient B is a Swiss male, who was diagnosed as KS based on the presence of hypogonadotropic hypogonadism and complete anosmia. The olfactory bulbs could not be visualised by MRI. Virilisation was achieved by testosterone therapy. At the age of 31, however, he had complete azoospermia that persisted after several months of hormonal treatment with gonadotropins.
Methods
Written informed consent was obtained from all the individuals who participated in the study.
Mutation analysis
Genomic DNAs were obtained from blood samples, by using a standard phenol chloroform extraction procedure. Each of the PROK2 four coding exons and flanking splice sites was PCR-amplified and sequenced using previously reported primers,12 and standard PCR and sequencing conditions. The mutations were confirmed on a second PCR product. The alleles from 100 control individuals of Turkish origin were analysed by using a denaturing high performance liquid chromatography standard procedure.
FISH analysis
Peripheral blood lymphocytes from patient B were cultivated for 72 h in medium 199 (Gibco-BRL). Prometaphase chromosomes were prepared by standard procedures, after thymidine synchronisation and BrdU incorporation. They were hybridised with BAC RP11-321A23 (The Wellcome Trust Sanger Institute) that encompasses the PROK2 gene at chromosome 3p13. DNA from the BAC was Texas red-labelled by nick translation. Chromosomes were visualised by DAPI counterstaining.
Analysis of polymorphic markers
Genotyping was carried out in patient B, with four polymorphic microsatellite markers that span a 8 Mb chromosomal region containing PROK2, namely D3S3697, D3S1261, D3S1284, and D3S3581. Standard procedures were used for PCR and automated fluorescent genotyping.
Results and discussion
In patient A, a biallelic missense mutation, c. 217C>T (p.R73C), was found in PROK2 exon 2. The mutation was also present in the heterozygous state in the patient's seemingly unaffected parents (see Figure 1). This mutation was not found in 200 alleles from ethnically matched control individuals. It produces a cysteine residue, which is expected to disrupt the formation of the protein disulphide bonds. Notably, the same mutation has already been reported in the heterozygous state in a KS sporadic case.12 No additional mutation was detected in the coding sequences of PROKR2, FGFR1, and KAL1 in patient A.
In patient B, a frameshift mutation, c.163delA (p.I55fsX1), creating a stop codon at the same codon position, was detected in PROK2 exon 2, in a seemingly homozygous state (see Figure 1). The parents' genetic status, however, could not be determined since the father died before this study. To rule out the possibility of a PROK2 deletion on one chromosome, we carried out a FISH analysis (see Methods) in the patient, which showed the presence of a fluorescent signal at both PROK2 chromosomal loci (data not shown), thus arguing against a large deletion. We then analysed in the patient four polymorphic markers spanning a 8 Mb interval containing PROK2 (see Methods), and found that he was carrying only one allelic form of each marker (data not shown). Since patient B is unlikely to have a large deletion of this chromosomal region, he must be homozygous for these polymorphic alleles. This genetic condition could be the consequence of either unrecognised parental consanguinity or unidisomy. We conclude that patient B is most probably homozygous for the c.163delA mutation too. No additional mutation was detected in the coding sequences of PROKR2, FGFR1, and KAL1. While this article was under review, the same PROK2 mutation was reported in the homozygous state in two brothers affected by KS, and their sister with isolated hypogonadotropic hypogonadism.15 The premature stop codon is expected to result in mRNA decay or a truncated peptide of 27 amino-acids in its mature form. This peptide was not able to activate the PROKR2 receptor produced by transfected CHO cells.15
The presence of PROK2 biallelic mutations in only two out of 320 patients analysed (273 sporadic cases and 47 familial cases), indicates that this genetic status is rare in KS. In addition, the present findings argue in favour of a digenic inheritance of the disease in patients carrying PROK2 mutations in the heterozygous state. Indeed, the same R73C mutation in PROK2 has been identified in the heterozygous12 and homozygous (this study) states, in two unrelated patients with the same disease phenotype. Moreover, only homozygous knockout mice for Prok2 have a phenotype that resembles KS, whereas heterozygous mice do not appear to have an abnormal phenotype.15, 16 Finally, the fact that some individuals heterozygous for PROK2 mutations are clinically unaffected argues against a dominant negative effect of these mutations. Therefore, we suggest that patients heterozygous for PROK2 mutations carry another disease-causing mutation in a different KS gene. Notably, the same prediction, based on similar findings, was made in patients heterozygous for PROKR2 mutations, and an additional KAL1 mutation has been identified in one of them.12 In the other patients heterozygous for PROKR2 or PROK2 mutations, no additional mutation has been found in the KS genes known to date. This suggests that the sought after mutations are located in one or several other genes. Indeed, barely 30% of all KS patients carry a mutation in KAL1, FGFR1, PROKR2, or PROK2, which indicates that still other genes underlying the disease remain undiscovered. Alternatively, the additional mutations might be located in as yet unexplored noncoding sequences of known KS genes.
References
Kallmann FJ, Schoenfeld WA, Barrera SE : The genetic aspects of primary eunuchoidism. Am J Ment Defic 1944; XLVIII: 203–236.
de Morsier G : Median cranioencephalic dysraphias and olfactogenital dysplasia. World Neurol 1962; 3: 485.
Naftolin F, Harris GW, Bobrow M : Effect of purified luteinizing hormone releasing factor on normal and hypogonadotropic anosmic men. Nature 1971; 232: 496–497.
Schwanzel-Fukuda M, Bick D, Pfaff DW : Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Mol Brain Res 1989; 6: 311–326.
Franco B, Guioli S, Pragliola A et al: A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991; 353: 529–536.
Legouis R, Hardelin J-P, Levilliers J et al: The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 1991; 67: 423–435.
Hardelin J-P, Levilliers J, del Castillo I et al: X chromosome-linked Kallmann syndrome: stop mutations validate the candidate gene. Proc Natl Acad Sci USA 1992; 89: 8190–8194.
Dodé C, Levilliers J, Dupont J-M et al: Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 2003; 33: 463–465.
Hardelin J-P, Julliard AK, Moniot B et al: Anosmin-1 is a regionally restricted component of basement membranes and interstitial matrices during organogenesis: implications for the developmental anomalies of X chromosome-linked Kallmann syndrome. Dev Dyn 1999; 215: 26–44.
Gonzalez-Martinez D, Kim SH, Hu Y et al: Anosmin-1 modulates fibroblast growth factor receptor 1 signaling in human gonadotropin-releasing hormone olfactory neuroblasts through a heparan sulfate-dependent mechanism. J Neurosci 2004; 24: 10384–10392.
Tsai P-S, Gill J : Mechanisms of disease: insights into X-linked and autosomal-dominant Kallmann syndrome. Nat Clin Pract Endocrinol Metab 2006; 2: 160–171.
Dodé C, Teixeira L, Levilliers J et al: Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genetics 2006; 2: 1648–1652.
Matsumoto S, Yamazaki C, Masumoto KH et al: Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci USA 2006; 103: 4140–4145.
Pitteloud N, Quinton R, Pearce S et al: Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest 2007; 117: 457–463.
Pitteloud N, Zhang C, Pignatelli D et al: Loss-of-function mutation in the prokineticin-2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA 2007; 104: 17447–17452.
Ng KL, Li JD, Cheng MY, Leslie FM, Lee AG, Zhou QY : Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science 2005; 308: 1923–1927.
Acknowledgements
We thank the patients and family members who participated in this study. This work was supported by a grant from Agence nationale pour la recherche (ANR-05-MRAR-027-01).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Leroy, C., Fouveaut, C., Leclercq, S. et al. Biallelic mutations in the prokineticin-2 gene in two sporadic cases of Kallmann syndrome. Eur J Hum Genet 16, 865–868 (2008). https://doi.org/10.1038/ejhg.2008.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.15
Keywords
This article is cited by
-
Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease
Human Genetics (2021)
-
Sexually dimorphic distribution of Prokr2 neurons revealed by the Prokr2-Cre mouse model
Brain Structure and Function (2017)
-
Point mutations in KAL1 and the mitochondrial gene MT-tRNAcys synergize to produce Kallmann syndrome phenotype
Scientific Reports (2015)
-
European Consensus Statement on congenital hypogonadotropic hypogonadism—pathogenesis, diagnosis and treatment
Nature Reviews Endocrinology (2015)
-
Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism
Nature Reviews Endocrinology (2012)
