
Abstract
Ferroptosis is a recently recognized form of regulated cell death. It is characterized morphologically by the presence of smaller than normal mitochondria with condensed mitochondrial membrane densities, reduction or vanishing of mitochondria crista, and outer mitochondrial membrane rupture. It can be induced by experimental compounds (e.g., erastin, Ras-selective lethal small molecule 3, and buthionine sulfoximine) or clinical drugs (e.g., sulfasalazine, sorafenib, and artesunate) in cancer cells and certain normal cells (e.g., kidney tubule cells, neurons, fibroblasts, and T cells). Activation of mitochondrial voltage-dependent anion channels and mitogen-activated protein kinases, upregulation of endoplasmic reticulum stress, and inhibition of cystine/glutamate antiporter is involved in the induction of ferroptosis. This process is characterized by the accumulation of lipid peroxidation products and lethal reactive oxygen species (ROS) derived from iron metabolism and can be pharmacologically inhibited by iron chelators (e.g., deferoxamine and desferrioxamine mesylate) and lipid peroxidation inhibitors (e.g., ferrostatin, liproxstatin, and zileuton). Glutathione peroxidase 4, heat shock protein beta-1, and nuclear factor erythroid 2-related factor 2 function as negative regulators of ferroptosis by limiting ROS production and reducing cellular iron uptake, respectively. In contrast, NADPH oxidase and p53 (especially acetylation-defective mutant p53) act as positive regulators of ferroptosis by promotion of ROS production and inhibition of expression of SLC7A11 (a specific light-chain subunit of the cystine/glutamate antiporter), respectively. Misregulated ferroptosis has been implicated in multiple physiological and pathological processes, including cancer cell death, neurotoxicity, neurodegenerative diseases, acute renal failure, drug-induced hepatotoxicity, hepatic and heart ischemia/reperfusion injury, and T-cell immunity. In this review, we summarize the regulation mechanisms and signaling pathways of ferroptosis and discuss the role of ferroptosis in disease.
Similar content being viewed by others

Facts
-
Ferroptosis is an iron- and ROS-dependent form of regulated cell death (RCD).
-
Ferroptosis is distinct from other forms of RCD at morphological, biochemical, and genetic levels.
-
Several molecules (e.g., VDAC2/3, glutathione peroxidase (GPX4), heat shock protein beta-1 (HSPB1), nuclear factor E2-related factor 2 (NRF2), NADPH oxidase (NOX), p53, and SLC7A11) regulate ferroptosis by directly or indirectly targeting iron metabolism and lipid peroxidation.
-
Misregulated ferroptosis has been implicated in multiple physiological and pathological processes such as cancer cell death, tissue injury, and T-cell immunity.
Open Questions
-
How does the downstream signaling or executor of iron-dependent ROS metabolism identify and distinguish ferroptosis from other types of RCD?
-
What controls the network of ferroptosis-signaling pathways?
-
What explains the cross-regulation between ferroptosis and other types of RCD?
-
How do mitochondrial dynamics and endoplasmic reticulum (ER) stressors affect ferroptosis?
-
What is the specific role of ferroptosis in human disease?
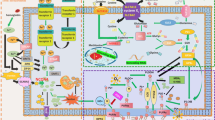
Ferroptosis is the term for a form of RCD that was recently coined in 2012 by the lab of Dr. Brent R Stockwell.1 According to their original study, ferroptosis is remarkably distinct from other types of RCD such as apoptosis, necroptosis, and autophagic cell death at morphological, biochemical, and genetic levels (Table 1). Multiple inducers and inhibitors of ferroptosis have been identified to affect accumulation of lipid peroxidation products and lethal reactive oxygen species (ROS) derived from iron metabolism (Table 2). Understanding the molecular mechanisms and signaling pathways (Figure 1) of ferroptosis may provide new diagnostic and therapeutic approaches to regulate cell survival and death in human disease.

Molecular mechanisms and signaling pathways of ferroptosis. (a) Core regulators of ferroptosis. (b–d) Roles of iron metabolism; (b), ROS metabolism (c), and the MAPK pathway (d) in ferroptosis
Discovery
Ferroptosis inducers were discovered before the notion of ferroptosis was invented. First identified as a ferroptosis inducer in 2003, erastin was found to be synthetic lethal with expression of the engineered mutant Ras oncogene in human foreskin fibroblasts (BJeLR), but not their isogenic primary counterparts.2 Ras-selective lethal small molecule (RSL)-3 and RSL5 were later identified in 2008 in another high-throughput small molecule-screening study that selectively killed BJeLR cells in a non-apoptotic manner.3 Inhibition of apoptosis, necrosis, necroptosis, and autophagy by small molecule inhibitors (e.g., Z-VAD-FMK, BOC-D-FMK, wortmannin, and necrostatin-1) cannot reverse RSL-induced cell death (Table 2).1 In contrast, antioxidants (e.g., vitamin E) and iron chelators (e.g., deferoxamine mesylate) block RSLs-induced cell death (Table 2).1 Thus, ferroptosis generally refers to an iron-dependent, non-apoptotic form of RCD.1
Morphology
Ferroptotic cancer cells are usually rounded up and detached in response to erastin.1, 2, 4 Ferroptotic cells exhibit changed mitochondrial morphology and cristae structure. Smaller than normal mitochondria with increased mitochondrial membrane density and reduction/vanishing of mitochondria crista have been observed in ferroptosis following erastin treatment in BJeLR cells.1, 2, 4 Induction of ferroptosis by genetic inactivation of GPX4 in immortalized fibroblasts and kidney tissue has been associated with outer mitochondrial membrane rupture, as observed using transmission electron microscopy.5 In contrast, the structural integrity of the nucleus is retained following erastin treatment in cancer cells.2 No nuclear condensation or chromatin margination are observed following erastin treatment in cancer cells.2 These morphological features help us distinguish ferroptosis from apoptosis, necroptosis, and autophagy (Table 1).
Inducer
Erastin
Cell death triggered by erastin is significantly inhibited by antioxidants (e.g., α-tocopherol, butylated hydroxytoluene, and β-carotene) and iron chelators (e.g., deferoxamine), suggesting that ROS- and iron-dependent signaling is required for erastin-induced ferroptosis (Figure 1a).1, 4 Six high-confidence genes regulating iron or mitochondrial fatty-acid metabolism are specifically required for erastin-induced ferroptosis.1 These genes include ribosomal protein L8, iron-responsive element-binding protein 2 (IREB2), ATP synthase F0 complex subunit C3, citrate synthase, tetratricopeptide repeat domain 35, and andacyl-CoA synthetase family member 2 (ACSF2).1 Activation of RAF/MEK/ERK signaling seems to be important for erastin-induced ferroptosis in tumor cells bearing oncogenic Ras.4 In vivo, piperazine erastin has been shown to have better stability and water solubility than erastin to inhibit cancer growth.6
One of the direct molecular targets of erastin is the mitochondrial voltage-dependent anion channel (VDAC) (Figure 1c).4 Erastin can directly bind to VDAC2/3 in BJeLR cells. Knockdown of VDAC2 and VDAC3, but not VDAC1, leads to erastin resistance.4 Furthermore, erastin has the ability to reduce glutathione (GSH) level by directly inhibiting cystine/glutamate antiporter system Xc− activity (Figure 1a),1 with activation of the ER stress response.7 This process could accelerate ROS accumulation during ferroptosis.
RSL3 and RSL5
Iron, ROS, and MEK are required for RSL3- and RSL5-induced ferroptosis in tumor cells bearing oncogenic Ras.3 VDAC2/3 is required for RSL5-, but not RSL3-induced ferroptosis.3 RSL3 is a direct inhibitor of GPX4, but not system Xc−.6 After binding to GPX4, RSL3 inactivates GPX4 to induce ROS production from lipid peroxidation (Figure 1a).6 Thus, at least two types of RSLs exist. Type I RSLs such as erastin and RSL5 can target upstream regulators (e.g., VDAC and system Xc−) to induce ferroptosis. Type II RSLs such as RSL3 can trigger ferroptosis by inhibiting downstream regulators (e.g., GPX4). The protein-synthesis inhibitor cycloheximide significantly inhibits RSL5-, but not RSL3-induced ferroptosis, indicating that protein synthesis is required for Type I RSL-induced ferroptosis.3
Buthioninesulfoximine
Buthioninesulfoximine (BSO) is an irreversible inhibitor of γ-glutamyl cysteine synthetase, the rate-limiting enzyme for GSH synthesis (Figure 1a). BSO inhibits GSH synthesis with decreased GPX activity and increased ROS levels, which results in ferroptosis in Ras-mutated cells.6
Acetaminophen
The reactive metabolite of acetaminophen has been identified as N-acetyl-p-benzoquinone imine, which causes GSH depletion and increases liver damage (Figure 1a). In addition to necrosis and apoptosis, acetaminophen induces ferroptosis in primary mouse hepatocytes, but not in HepG2 liver cancer cell lines.8 The protective effect of ferrostatin-1 against acetaminophen-induced cell death does not occur from the reduced metabolism of acetaminophen or GSH depletion.8
FIN
In a larger screening to find ferroptosis-inducing compounds, a series of small molecule inducers, namely ferroptosis-inducing agents (FINs), were discovered.9, 10 Each of these additional FINs generates ROS, whereas BHT strongly suppresses this lethal ROS-induced cell death.6 In the LC-MS-based GPX4 assay, cells treated with any of the seven DPI family members (DPI7, DPI10, DPI12, DPI13, DPI17, DPI18, and DPI19) are unable to detect reduced PC-OOH levels, which is a specific substrate of GPX4.6 Like RSL3, these FIN compounds (class II FINs) directly inhibit GPX4 activity without GSH depletion (Figure 1a).6 Class I FINs such as DPI2 enacts the same mechanism as erastin and BSO to inhibit GPX4 by GSH depletion.6
Lanperisone
Lanperisone is a modified form of tolperisone, which was developed as a muscle relaxant.11 Lanperisone can selectively kill K-Ras-mutant mouse embryonic fibroblasts through the induction of ROS, which is mediated through iron and Ras/RAF/MEK/ERK signaling (Figure 1d). In vivo, lanperisone also showed efficacy against tumor growth in a K-Ras-driven mouse model of lung cancer.12 The exact mechanism responsible for lanperisone-induced ROS generation is not known, but preliminary results suggest that it occurs through the perturbation of voltage-gated ion channels.11
Sulfasalazine
Sulfasalazine is broadly used to treat chronic inflammation in the gut, joints, and retina. In addition to inhibiting the NF-κB signaling pathway, sulfasalazine inhibits the system Xc− transporter (Figure 1a).13 Given that the disruption of system Xc−-mediated cystine uptake by erastin is sufficient to induce ferroptosis, treatment of cancer cells (e.g., BJeLR and HT1080) with sulfasalazine also triggers ferroptosis.1, 7
Sorafenib
Sorafenib induces ferroptosis in certain cancer cells such as hepatocellular carcinoma (HCC) cells.14, 15 Sorafenib-induced ferroptosis occurs independent from the oncogenic status of Ras, RAF, PIK3CA, and p53.16 However, the expression of Rb (the prototype tumor suppressor gene) and NRF2 could inhibit sorafenib-induced ferroptosis in HCC.14, 17 The mechanism and action of sorafenib in ferroptosis may depend on the inhibition of system Xc− function, but not on GPX4 activity (Figure 1a).7 This process is associated with upregulated ER stress.7 Structure activity relationship analysis of 87 sorafenib analogs further indicates that sorafenib inhibits system Xc− activity via a non-kinase target.7
Artesunate
Artesunate selectively induces ferroptosis in K-Ras-mutant pancreatic ductal adenocarcinoma (PDAC) cell lines, but not human pancreatic ductal epithelial cells or wild-type K-Ras PDAC cells.18 This process can be blocked by the iron chelator deferoxamine, whereas it is enhanced by exogenous lysosomal form of iron.18 In addition to iron chelator deferoxamine, the ROS inhibitors (e.g., trolox and ferrostatin-1), but not necrostatin-1, significantly inhibit artesunate-induced ferroptosis in PDAC cells.18 Another study shows that 10 artemisinin derivatives, including artesunate, altered numerous iron-related gene mRNA levels, which contributes to cell death in cancer cells.19
Signaling pathway
Iron metabolism and lipid peroxidation signaling are increasingly recognized as central mediators of ferroptosis.20 In addition, activation of the mitogen-activated protein kinase (MAPK) pathway contributes to ferroptotic cancer cell death.
Iron
Excessive iron contributes to ferroptosis through producing ROS by Fenton reaction (Figure 1a). Circulating iron exists in the form of ferric iron (Fe3+) by binding to transferrin. Fe3+ is imported into cells through the membrane protein transferrin receptor 1 (TFR1) and then locates in the endosome. In the endosome, Fe3+ is reduced to ferrous iron (Fe2+) by the ferrireductase activity of STEAP3. Finally, divalent metal transporter 1 (DMT1, also termed SLC11A2) mediates the release of Fe2+ from the endosome into a labile iron pool in the cytoplasm. Excess iron is stored in ferritin, an iron storage protein complex including ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1). Iron export is mediated by the membrane protein ferroportin (an iron efflux pump, also termed SLC11A3), which can oxidize Fe2+ to Fe3+.
Ferroptosis-sensitive cells with Ras mutation have increased TFR1 and decreased ferritin (FTL and FTH1) expression compared with ferroptosis-resistant cells.3 This suggests that increased iron uptake and reduced iron storage may contribute to iron overload during ferroptosis. Indeed, decreased iron overload by iron chelators (e.g., deferoxamine, desferrioxamine mesylate, ciclopirox olamine) inhibits erastin-mediated ferroptosis, whereas supplying exogenous sources of iron (e.g., ferric ammonium citrate, ferric citrate, and iron chloridehexahydrate) enhances erastin-induced death.1, 3 More direct evidence for iron-dependent ferroptosis comes from the knockdown of IREB2, a master transcription factor of iron metabolism. Suppression of IREB2 expression by RNAi significantly increases iron metabolism-associated gene expression (e.g., F-box and leucine-rich repeat protein 5, iron-sulfur cluster assembly enzyme, FTH1, and FTL) and limits erastin-induced ferroptosis.1, 3 Thus, the cellular systems involved in the uptake and utilization of iron are required for the induction of ferroptosis.
ROS
The ROS origin of ferroptosis induction may involve multiple sources (Figure 1c). In addition to iron-mediated ROS production by Fenton reaction, nicotinamide adenine dinucleotide phosphate (NADPH)-dependent lipid peroxidation and GSH depletion are also important for the induction of ferroptosis.1, 6 Inactivation of GPX4 by GSH depletion triggers ferroptosis by accumulation of ROS production from lipid peroxidation.6 Mitochondrial fatty-acid metabolism provides the specific lipid precursor required for ferroptosis. In particular, ACSF2 and CS are required for mitochondrial fatty-acid metabolism in ferroptosis. Knockdown of ACSF2 and CS inhibits erastin-induced ferroptosis.1 In addition to glucose metabolism, lipid production can generate from the conversion of glutamine to α-ketoglutarate (Figure 1c). This process can be blocked by the small molecule transaminase inhibitor aminooxyacetic acid.
ROS can react with the polyunsaturated fatty acids (PUFAs) of lipid membranes and induce lipid peroxidation. Two lipid metabolism-associated genes (lysophosphatidylcholine acyltransferase 3 (LPCAT3) and acyl-CoA synthetase long-chain family member 4 (ACSL4)) have been identified to promote RSL3− and DPI7 (also known as ML162)−, but not erastin-induced ferroptosis by using haploid genetic screening in KBM7 cells.21 The depletion of arachidonic acid (AA) and other PUFAs, following GPX4 inactivation, is required for the execution of ferroptosis.5, 22 ACSL4 acylates AA and LPCAT3 catalyzes the acylated AA into membrane phospholipids. Thus, suppression of ACSL4 and LPCAT3 may decrease the oxidization of a number of sensitive fatty acids in the membrane. In contrast, GPX4-deficient cells exhibit enriched oxidized membranes with AA, which contributes to ferroptosis.5 The release of AA mediators, including 5-hydroxyeicosatetraenoic acid (HETE), 11-HETE, and 15-HETE, but not 12-HETE in the medium of GPX4−/− cells, has been specifically observed following ferroptosis stimuli (e.g., RSL3 and erastin), but not apoptosis stimuli (e.g., TNFα and staurosporine).5 Treatment with 5-, 12-, and 15-hydroperoxyeicosatetraenoic acid (HPETE) accelerates the ferroptotic process.5 Pharmacological inhibition of sterol carrier protein 2, a lipid transport protein for mitochondria, transiently rescues GPX4−/− cells from ferroptosis. The priming of lethal lipid signals from GPX4−/− cells appears to occur outside the mitochondrial matrix.5 Thus, ferroptosis is triggered by extra-mitochondrial lipid peroxidation, which is associated with oxidized lipid mediator release.
Another study using erastin-resistant DU-145 cancer cells indicates that upregulation of AKR1C family genes, including AKR1C1-3, is associated with ferroptosis resistance.7 AKR1C1-3 catalyzes the oxidation of 4-hydroxynonenal into various PUFAs. These erastin-resistant DU-145 cells also exhibit resistance to sulfasalazine, sorafenib, and RSL3, suggesting that the resistance is unlikely to be due to increased system Xc− activity.
Acetyl-CoA carboxylase alpha (encoding ACC1), the rate-limiting step in fatty-acid synthesis, seems to be not required for ferroptosis in certain cells.21 Knockdown of ACC1 by RNAi or inhibition of ACC1 by 5-tetradecyloxy-2-furonic acid did not affect RSL3- and erastin-induced ferroptosis.21 PUFAs such as AA and linolenic acid are substrates of lipoxygenases (LOX). LOX oxidizes PUFAs to their hydroperoxyl intermediates, including HPETE, in the case of AA. PUFAs decrease when lipid peroxidation increases. When AA is the substrate, different LOX isozymes can add a hydroperoxyl group at carbons 5, 12, or 15, and therefore are designated as 5-, 12- or 15-LOX. However, 12/15-LOX does not appear to be required for ferroptosis.5
MAPK
The mammalian family of MAPKs mainly includes ERK, p38, and c-Jun NH2-terminal kinase (JNK). Blocking the Ras/Raf/MEK/ERK pathway inhibits erastin-induced ferroptosis in Ras-mutated cancer cells.4 However, JNK and p38, but not ERK, appear important for erastin-induced cell death in leukemia cells.23 SP600125 (an inhibitor of JNK phosphorylation) and SB202190 (an inhibitor of p38 activation) decrease the cytotoxicity induced by erastin in HL-60 cells.23 These findings indicate that ferroptotic responses associated with a certain MAPK module can be cell type-specific (Figure 1d).
Molecular biology
Positive regulators of ferroptosis
Vdac2/3
VDAC2/3, but not VDAC1, has a positive role in ferroptosis because knockdown of VDAC2/3, but not VDAC1, inhibits erastin-induced ferroptosis in Ras-mutated cells (Figure 1c).4 VDAC2/3 has been found to be a direct target of erastin via affinity purification assay.4 Cells with more VDAC protein are more sensitive to erastin.4 Erastin decreases the rate of NADH oxidation in isolated yeast mitochondria expressing a single mouse VDAC isoform4 and increases the permeance of NADH into liposomes containing human VDAC2.24 Erastin can enhance oxidative mitochondrial metabolism and limit aerobic glycolysis by disrupting the interaction between VDAC and tubulin in human liver cancer cells (e.g., HepG2), suggesting a potential role of energy metabolism and cytoskeleton in the modification of ferroptosis.25
Ras
Erastin exhibits gene-selective lethality in Ras-mutant cells, including H-Ras-mutant engineered cells, N-Ras-mutant HT1080 cells, and K-Ras-mutant Calu-1 cells.4 However, induction of ferroptosis may exist in a both Ras-dependent and -independent manner. Artesunate induces pancreatic cancer death in a Ras-dependent manner,18 whereas it induces leukemia cell death in a Ras-independent manner.23 Indeed, several normal Ras wild-type cells such as kidney tubule cells, T cells, and fibroblasts are sensitive to erastin.5, 22, 26, 27, 28 Even in some cases, overexpression of mutant Ras in rhabdomyosarcoma cells (e.g., RMS13 cells) promotes ferroptosis resistance to erastin and RLS3.29
TFR1
Compared with ferroptosis-resistant cells (e.g., BJ cells), the expression of TFR1 is upregulated in ferroptosis-sensitive cells (e.g., BJeLR cells).3 In contrast, the expression of FTH1 and FTL is downregulated in ferroptosis-sensitive cells (e.g., BJeLR cells), indicating that iron storage also affects ferroptosis.3 Knockdown of TFR1 by shRNA inhibits erastin-induced ferroptosis in BJeLR cells, confirming that inhibition of iron uptake prevents ferroptosis.3 Transferrin and glutamine are two important components of full fetal bovine serum, which induces ferroptosis in fibroblasts upon amino acid starvation.30
NOX
The NOX protein family transfers electrons across biological membranes to reduce oxygen to superoxide. The canonical NOX inhibitor diphenyleneiodonium and the NOX1/4-specific inhibitor GKT137831 partly inhibit erastin-induced ferroptosis in Calu-1 and HT1080 cells (Figure 1c).1 The pentose phosphate pathway (PPP) is a metabolic pathway parallel to glycolysis that generates NADPH and pentoses. Pharmacological inhibition of PPP by 6-aminonicotinamde (Figure 1c) or knockdown of two PPP enzymes (glucose-6-phosphate dehydrogenase and phosphoglycerate dehydrogenase) also partly prevents erastin-induced ferroptosis in Calu-1 cells.1
p53
Activation of p53 has been found to be required for ferroptosis in certain cancer cells.27 This process depends on direct transcriptional inhibition of SLC7A11, a key component of system Xc− (Figure 1a) (discussed later). Moreover, p533KR (an acetylation-defective mutant) is responsible for the inhibition of SLC711A expression (Figure 1a), but not other known p53 target genes (e.g., p21 and BAX) involved in antiproliferative and pro-apoptotic activity.27 p533KR mice lacking cell-cycle arrest, apoptosis, or senescence still exhibit tumor suppression function.31 This tumor suppressor function of p533KR is dependent on the induction of ferroptosis.27 Ferroptosis also mediates hyperactive p53 signaling in the promotion of embryonic lethality.27 The E3 ubiquitin ligase murine double minute-2 (MDM2) regulates the proteasomal degradation of p53. Ferrostatin-1 alone cannot prevent knockdown of MDM2 by RNAi-induced cell death,32 indicating that mixed cell death types are responsible for p53-induced death.
CARS
Cysteinyl-tRNA synthetase (CARS) is a positive regulator of ferroptosis upon cystine deprivation.33 Knockdown of CARS inhibits erastin-induced ferroptosis, whereas overexpression of CARS enhances erastin sensitivity in several types of cancer cells (Figure 1c).33 However, loss of CARS cannot prevent RSL3-, FIN56-, and BSO-induced cell death, suggesting that CARS regulates ferroptosis at the cysteine biosynthesis level.33
Negative regulators of ferroptosis
GPX4
GPX4 converts reduced GSH to oxidized glutathione (GSSG) while reducing lipid hydroperoxides to their corresponding alcohols or free hydrogen peroxide to water. Treatment with erastin or BSO can deplete GSH and GSSG and increase NADPH oxidation and lysophosphatidylcholines (an indicator of generation of ROS from lipid).6 In contrast, GSH and N-acetylcysteine (a GSH biosynthetic precursor) prevent erastin lethality in U2OS cells (Figure 1a).6 However, RSL3 can generate ROS in the absence of GSH depletion.6 GPX4 is a direct target of RLS3. Knockdown of GPX4 induces ferroptosis in an iron-, MEK-, and ROS-dependent manner, whereas overexpression of GPX4 leads to resistance to RSL3.6 Erastin is also able to cause GPX4 degradation in several types of cancer cells, suggesting that the protein degradation pathway is involved in ferroptosis.23
The function of ferroptosis in vivo is confirmed by using GPX4 conditional or inducible knockout mice. Inducible knockout of GPX4 in the kidney leads to acute renal failure, which can be rescued by ferrostatin-1 and necrostatin-1 (a necroptosis inhibitor).5 Necrostatin-1 may have an off-target effect in the inhibition of ferroptosis in the kidney.5 In the presence or absence of lymphocytic choriomeningitis virus infection, fewer CD8+ T cells have been observed in the spleens and lymph nodes of mice with conditional knockout of GPX4 in T cells by Cd4-Cre.26 GPX4−/− T cells rapidly accumulate lipid peroxides and die by ferroptosis, but not other types of RCD.26 Inducible neuron-specific GPX4 knockout mice have neuronal loss in the brain partly due to induction of ferroptosis.34 In contrast, mice with deletion of GPX4 in hematopoietic cells develop anemia due to induction of RIP3-dependent necroptosis, but not apoptosis and ferroptosis.35 These results indicate context-dependent functions of the GPX4 in cell death.
System Xc−
System Xc− is responsible for maintaining redox homeostasis by importing cystine, where it is then reduced to cysteine and used to synthesize the major antioxidant GSH. Inhibiting system Xc− with sulfasalazine can trigger ferroptosis, whereas increasing cystine uptake into cells by β-mercaptoethanol inhibits erastin-induced ferroptosis in HT1080 cells (Figure 1a).1 System Xc− structurally consists of SLC7A11 and SLC3A2. The upregulation of SLC7A11 by erastin is not dependent on iron and ROS.1 Suppression of SLC7A11 expression by RNAi increases the anticancer activity of erastin, whereas overexpression of SLC7A11 by gene transfection diminishes erastin-induced ferroptosis.1 In contrast, RSL3-mediated ferroptosis is system Xc−-independent.1 Several ER stress markers such as phosphorylation of eIF2α and ATF4 protein upregulation has been observed in ferroptosis following treatment with erastin, sulfasalazine, and sorafenib.7 As discussed above, p53 is a positive regulator of ferroptosis in certain cancer cells by inhibition of SLC7A11 expression, therefore inhibiting system Xc− activity.7
HSPB1
HSPB1 expression is remarkably induced by the transcriptional factors heat shock factor-1 (HSF-1) after erastin treatment in several human cancer cells.36 Inhibition of HSF-1-dependent HSPB1 expression increases, whereas overexpression of HSPB1 inhibits erastin-induced ferroptosis.36 The phosphorylation of HSPB1 is important for its function in the regulation of actin dynamics and iron uptake. HSPB1 phosphorylation is increased by protein kinase C (PKC) following erastin treatment in HeLa cells, which facilitates ferroptotic resistance through blocking cytoskeleton-mediated iron uptake and subsequent ROS production (Figure 1b).36
NRF2
NRF2 has an anti-ferroptosis role in HCC cells.17 p62 interacts with the NRF2-binding site of Kelch-like ECH-associated protein 1 (Keap1) and competitively inhibits Keap1-NRF2 interaction, which is responsible for NRF2 protein stability following treatment with FINs (e.g., erastin, sorafenib, and BSO).17 Upregulated NRF2 protein promotes transcription of genes encoding antioxidant proteins (e.g., quinone oxidoreductase 1 and heme oxygenase-1 (HO-1)) and iron metabolism proteins (e.g., FTH1) in ferroptosis.17 Knockdown of NRF2 and these NRF2-targeted genes accelerates erastin or sorafenib-induced ferroptosis in HCC cells.17 In contrast, induction of HO-1 expression by erastin may promote cell death in HT1080 and fibroblasts, suggesting that HO-1 has a dual role in ferroptosis.37
Measuring methods
In vitro
Cell viability
Four methods of cell viability measurement have been reported in the evaluation of ferroptosis: calceinacetoxymethyl ester (Calcein AM) viability assay,38 alamarBlue viability assay,39 trypan blue assay,40 and Cell Counting Kit-8 assay.23
Iron level
Phen Green SK (PGSK) is a cell membrane-permeable dye used to monitor the intracellular iron level in living cells via flow cytometry or confocal microscopy. It shows a diminished green autofluorescence of PGSK upon binding to sufficient cellular iron following erastin treatment.3 The Iron Assay Kit provides a simple convenient means of measuring ferrous and/or ferric ions in samples.17
ROS level
Erastin-treated HT1080 cells are accompanied by a prolonged period of lipid peroxidation detection by C11-BODIPY probe and shift the fluorescence from red to green, whereas ferroptosis inhibitors restrict this shift. In contrast, MitoSOX (a mitochondrial ROS probe) is not changed following erastin treatment.1 In addition, GSH depletion, glutamate release, and NADPH and cystine uptake assay can be used to monitor ferroptosis. GSH depletion and inhibition of glutamate release are observed in erastin- and sorafenib-induced ferroptosis.6, 7 Decreased cystine uptake and increased NADPH oxidation capability are associated with increased ferroptosis.6
In vivo
Prostaglandin-endoperoxide synthase (PTGS) is the key enzyme in prostaglandin biosynthesis. There are two isozymes of PTGS: a constitutive PTGS1 and an inducible PTGS2. PTGS2 encoding cyclooxygenase-2 (COX-2) is significantly upregulated after treatment with RSL3 and erastin in mice.6 Upregulated PTGS2 only indicates ferroptosis onset and does not affect ferroptosis development.6 Assessment of ferroptosis-associated genes/proteins in tissue and iron and GSH level in serum may be an alternative approach to monitor ferroptosis in vivo.
Inhibitor
Several ferroptosis inhibitors are listed in Table 2. Most of them are antioxidants or iron chelators. Here, we discuss three major compounds with specific anti-ferroptotic activity.
Ferrostatin
First-generation ferrostatin is termed ferrostatin-1 and acts to inhibit erastin- and RSL3-induced ferroptosis in HT1080 cells.1 The activity of ferrostain-1 depends on the primary aromatic amine, which specifically inhibits accumulation of ROS from lipid oxidation.1 Compared with ferrostatin-1, second- (termed SRS 11–92) and third-generation ferrostatins (termed SRS 16–86) have increased plasma and metabolic stability and significantly protect against tissue injury (e.g., acute kidney injury and ischemia-reperfusion injury) in vivo (Figure 1a).22, 28
Liproxstatin-1
Liproxstatin-1 prevents ROS accumulation and cell death in GPX4−/− cells (Figure 1a).5 Moreover, liproxstatin-1 inhibits FINs (e.g., erastin, RSL3, and BSO)-induced ferroptosis in vitro.5 Intraperitoneal administration of liproxstatin-1 (10 mg/kg) in inducible GPX4−/− mice prolongs animal survival in response to kidney injury.5 Liproxstatin-1 also protects against ischemia/reperfusion-induced liver injury in mice.5
Zileuton
Zileuton is an orally active specific inhibitor for 5-LOX, but not 12/15-LOX (Figure 1c). Zileuton provided significant protection from glutamate- and erastin- induced ferroptosis in HT22 cells (a mouse hippocampal cell line) by inhibition of cytosolic ROS production.41
Diseases
Cancer
NCI-60 is a panel of 60 diverse human cancer cell lines from eight diverse tissue types used by the US National Cancer Institute Developmental Therapeutics Program. Among them, kidney and leukemia cancer cells are more sensitive to erastin compared with cancer cells from the other six tissues (lung, colon, central nervous system, melanocytes, ovary, and breast).6 Furthermore, the anticancer activity of erastin was tested in 117 cancer cells and no correlation between Ras mutation and erastin potency was identified.6 Besides exerting single effects, erastin also enhances chemotherapy drugs such as temozolomide, cisplatin, cytarabine/ara-C, and doxorubicin/adriamycin) in certain cancer cells.23, 42, 43 Induction of ferroptosis by FDA-approved drugs such as sorafenib, sulfasalazine and artesunate holds great potential for cancer therapy. In vivo, erastin, piperazine erastin, and RSL3 prevented tumor growth in a xenograft model.6, 36 However, the role of ferroptosis in tumorigenesis still remains unclear.
Neurotoxicity
Inhibition of ferroptosis by ferrostatin-1 protects organisms from glutamate-induced neurotoxicity in a rat organotypic hippocampal slice culture model.1 Fms-like tyrosine kinase 3 (FLT-3, also termed CD135) is a cytokine receptor, which is important for the normal development of hematopoietic stem cells and progenitor cells. Potent inhibitors for FLT-3 and its downstream signaling molecule phosphoinositide 3-kinase α (p110a) can suppress ROS production and lipid peroxidation to block ferroptotic cell death in neurons44 (Figure 1c). Inhibition of ferroptosis by ferrostatins (ferrostatin-1 and SRS 11–92) also restored the number of healthy neurons in a Huntington’s disease model.22 Periventricular leukomalacia usually refers to the death of developing oligodendrocytes (OLs). Ferrostatins (e.g., ferrostatin-1 and SRS 11–92) remarkably protect OLs from cystine deprivation.22
AKF
Ferrostatins (e.g., ferrostatin-1 and SRS 11–92) had a protective role to prevent lethality of in a model of acute injury of freshly isolated renal tubules,22 implicating ferroptosis-mediated cell death in acute kidney failure (AKF). Inhibition of ferroptosis by third-generation ferrostatins (SRS 16–86) limits acute ischemia-reperfusion injury and oxalate nephropathy-related AKF.28 Similarity, inducible knockout of GPX4 in the kidney leads to ferroptosis, which contributes to AKF in mice.5
Liver injury
Acetaminophen overdoses are currently the most frequent cause of acute liver failure. Acetaminophen has been demonstrated to induce ferroptosis in primary liver cells and ferroptosis inhibitors such as ferrostain-1 can inhibit acetaminophen-induced death.8 Ferroptosis is also implicated in hepatic ischemic damage in mice. Ischemia/reperfusion-induced liver injury can be ameliorated in mice by the ferroptosis inhibitor liproxstatin-1.5
Hear injury
Inhibiting glutaminolysis and ferroptosis by compound 968, DFO, or ferrastatin-1 limits ischemia/reperfusion-induced heart injury ex vivo.30
Conclusion & perspective
Several molecules have recently been identified to regulate ferroptosis by directly or indirectly targeting iron metabolism and lipid peroxidation. These so-called ferroptosis regulators are also implicated in other types of RCD. Thus, the most important objective in the study of ferroptosis is to identify the downstream signaling pathways or executors of iron-dependent ROS metabolism to distinguish ferroptosis from other types of RCD.
Induction of ferroptosis by drugs has been shown to inhibit cancer cell growth in both a Ras-dependent and -independent manner, suggesting that cancer cells display genetic heterogeneity in the timing of the ferroptotic response. Further definition of the genotype-selective activity of ferroptosis in cancer and the mechanisms involved will be important to guide ferroptosis-based therapeutic intervention. Ferroptosis has an important role in sterile inflammatory conditions such as tissue acute injury, ischemic-reperfusion injury, and neurotoxicity. An improved understanding of the role of ferroptosis in cancer and injury-associated diseases will create a new opportunity for diagnosis and therapeutic intervention.
Abbreviations
- AA:
-
arachidonic acid
- ACC1:
-
acetyl-CoA carboxylase alpha
- ACSF2:
-
andacyl-CoA synthetase family member 2
- ACSL4:
-
acyl-CoA synthetase long-chain family member 4
- AOA:
-
aminooxyacetic acid
- AKF:
-
acute kidney failure
- ATP5G3:
-
ATP synthase F0 complex subunit C3
- BSO:
-
buthionine sulfoximine
- BHT:
-
butylated hydroxytoluene
- CARS:
-
cysteinyl-tRNA synthetase
- CHAC1:
-
cation transport regulator-like protein 1
- COX-2:
-
cyclooxygenase-2
- CS:
-
citrate synthase
- CSSG:
-
cysteine-glutathione disulfide
- DAMP:
-
damage-associated molecular pattern molecule
- DMT1:
-
divalent metal transporter 1
- ER:
-
endoplasmic reticulum
- Fe3+:
-
ferric iron
- Fe2+:
-
ferrous iron
- FIN:
-
ferroptosis-inducing agents
- FLT-3:
-
fms-like tyrosine kinase 3
- FTH1:
-
ferritin heavy chain 1
- FTL:
-
ferritin light chain
- GPX4:
-
glutathione peroxidase 4
- GSH:
-
glutathione
- HMGB1:
-
high mobility group box 1
- HD:
-
Huntington’s disease
- HETE:
-
hydroxyeicosatetraenoic acid
- HO-1:
-
heme oxygenase-1
- HPETE:
-
hydroperoxyeicosatetraenoic acid
- HSF-1:
-
heat shock factor-1
- HSPB1:
-
heat shock protein beta-1
- HCC:
-
hepatocellular carcinoma
- IREB2:
-
iron-responsive element-binding protein 2
- JNK:
-
c-Jun NH2-terminal kinase
- Keap1:
-
Kelch-like ECH-associated protein 1
- LOX:
-
lipoxygenases
- LPCAT3:
-
lysophosphatidylcholine acyltransferase 3
- MAPK:
-
mitogen-activated protein kinase
- MDM2:
-
murine double minute-2
- MEFs:
-
mouse embryonic fibroblasts
- NADPH:
-
nicotinamide adenine dinucleotide phosphate
- NAPQI:
-
N-acetyl-p-benzoquinone imine
- NOX:
-
NADPH oxidase
- NRF2:
-
nuclear factor erythroid 2-related factor 2
- OLs:
-
oligodendrocytes
- PDAC:
-
pancreatic ductal adenocarcinoma
- PGSK:
-
phen green SK
- PKC:
-
protein kinase C
- PPP:
-
pentose phosphate pathway
- PTGS:
-
prostaglandin-endoperoxide synthase
- PUFAs:
-
polyunsaturated fatty acids
- PVL:
-
periventricular leukomalacia
- RCD:
-
regulated cell death
- ROS:
-
reactive oxygen species
- RPL8:
-
ribosomal protein L8
- RSLs:
-
Ras-selective lethal small molecules
- SCP2:
-
sterol carrier protein 2
- TFR1:
-
transferrin receptor 1
- TTC35:
-
tetratricopeptide repeat domain 35
- VDAC:
-
mitochondrial voltage-dependent anion channel.
References
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149: 1060–1072.
Dolma S, Lessnick SL, Hahn WC, Stockwell BR . Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003; 3: 285–296.
Yang WS, Stockwell BR . Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008; 15: 234–245.
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007; 447: 864–868.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014; 16: 1180–1191.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014; 156: 317–331.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014; 3: e02523.
Lorincz T, Jemnitz K, Kardon T, Mandl J, Szarka A . Ferroptosis is Involved in Acetaminophen Induced Cell Death. Pathol Oncol Res 2015; 21: 1115–1121.
Yang WS, Shimada K, Delva D, Patel M, Ode E, Skouta R et al. Identification of simple compounds with microtubule-binding activity that inhibit cancer cell growth with high potency. ACS Med Chem Lett 2012; 3: 35–38.
Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett 2012; 22: 1822–1826.
Sakitama K, Ozawa Y, Aoto N, Tomita H, Ishikawa M . Effects of a new centrally acting muscle relaxant, NK433 (lanperisone hydrochloride) on spinal reflexes. Eur J Pharmacol 1997; 337: 175–187.
Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci USA 2011; 108: 8773–8778.
Gout PW, Buckley AR, Simms CR, Bruchovsky N . Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 2001; 15: 1633–1640.
Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett 2015; 356: 971–977.
Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer 2013; 133: 1732–1742.
Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res 2014; 34: 6417–6422.
Sun X, Ou Z, Chen R, Niu X, Chen, Kang R et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2015 Sep 24. doi: 10.1002/hep.28251. [Epub ahead of print].
Nils Eling, Lukas Reuter, Hazin John, Hamacher-Brady Anne, Brady NR . Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015; 2: 517–532.
Ooko E, Saeed ME, Kadioglu O, Sarvi S, Colak M, Elmasaoudi K et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015; 22: 1045–1054.
Dixon SJ, Stockwell BR . The role of iron and reactive oxygen species in cell death. Nat Chem Biol 2014; 10: 9–17.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 2015; 10: 1604–1609.
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 2014; 136: 4551–4556.
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2015 May 26. doi:10.1080/23723556.2015.1054549. [Epub ahead of print].
Bauer AJ, Gieschler S, Lemberg KM, McDermott AE, Stockwell BR . Functional model of metabolite gating by human voltage-dependent anion channel 2. Biochemistry 2011; 50: 3408–3410.
Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM et al. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem 2013; 288: 11920–11929.
Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M . T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med 2015; 212: 555–568.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015; 520: 57–62.
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA 2014; 111: 16836–16841.
Schott C, Graab U, Cuvelier N, Hahn H, Fulda S . Oncogenic RAS mutants confer resistance of RMS13 rhabdomyosarcoma cells to oxidative stress-induced ferroptotic cell death. Front Oncol 2015; 5: 131.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X . Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 2015; 59: 298–308.
Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149: 1269–1283.
Thomasova D, Bruns HA, Kretschmer V, Ebrahim M, Romoli S, Liapis H et al. Murinedouble minute-2 prevents p53-overactivation-related cell death (podoptosis) of podocytes. J Am Soc Nephrol 2014; 26: 1513–1523.
Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR . Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ 2015 Jul 17. doi: 10.1038/cdd.2015.93. [Epub ahead of print].
Chen L, Hambright WS, Na R, Ran Q . Ablation of ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem 2015; 290: 28097–28106.
Canli Ö, Alankuş YB, Grootjans S, Vegi N, Hültner L, Hoppe PS et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2015 Oct 13. pii: blood-2015-06-654194. [Epub ahead of print].
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015; 34: 5617–5625.
Kwon MY, Park E, Lee SJ, Chung SW . Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015; 6: 24393–24403.
Wang XM, Terasaki PI, Rankin GW Jr, Chia D, Zhong HP, Hardy S . A new microcellular cytotoxicity test based on calcein AM release. Hum Immunol 1993; 37: 264–270.
Page B, Page M . Sensitive colorimetric cytotoxicity measurement using alarmar blue. Oncol Rep 1995; 2: 59–61.
Avelar-Freitas BA, Almeida VG, Pinto MC, Mourao FA, Massensini AR, Martins-Filho OA et al. Trypan blue exclusion assay by flow cytometry. Braz J Med Biol Res 2014; 47: 307–315.
Liu Y, Wang W, Li Y, Xiao Y, Cheng J, Jia J . The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol Pharm Bull 2015; 38: 1234–1239.
Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y . Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol Rep 2015; 33: 1465–1474.
Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu MC, Chang SS et al. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin Cancer Res 2013; 19: 845–854.
Kang Y, Tiziani S, Park G, Kaul M, Paternostro G . Cellular protection using Flt3 and PI3Kalpha inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat Commun 2014; 5: 3672.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009 16: 3–11.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012 19: 107–120.
Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 2005; 12: 1463–1467.
Acknowledgements
We apologize to the researchers who were not referenced owing to space limitations. We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript. This work was supported by the National Institutes of Health of the USA (R01CA160417 and R01GM115366 to DT), The National Natural Science Foundation of China (31171229 and U1132005 to XS), and a Science and Information Technology of Guangzhou Key Project (201508020258 and 201400000003/4 to XS).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Rights and permissions
About this article

Cite this article
Xie, Y., Hou, W., Song, X. et al. Ferroptosis: process and function. Cell Death Differ 23, 369–379 (2016). https://doi.org/10.1038/cdd.2015.158
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.158
This article is cited by
-
TfR1 mediated iron metabolism dysfunction as a potential therapeutic target for osteoarthritis
Arthritis Research & Therapy (2024)
-
TrkA promotes MDM2-mediated AGPS ubiquitination and degradation to trigger prostate cancer progression
Journal of Experimental & Clinical Cancer Research (2024)
-
MiR-134-3p targets HMOX1 to inhibit ferroptosis in granulosa cells of sheep follicles
Journal of Ovarian Research (2024)
-
The microRNA-211-5p/P2RX7/ERK/GPX4 axis regulates epilepsy-associated neuronal ferroptosis and oxidative stress
Journal of Neuroinflammation (2024)
-
Exosomes derived from BMSCs enhance diabetic wound healing through circ-Snhg11 delivery
Diabetology & Metabolic Syndrome (2024)
