
Abstract
Cellular stress defense mechanisms have evolved to maintain homeostasis in response to a broad variety of environmental challenges. Stress signaling pathways activate multiple cellular programs that range from the activation of survival pathways to the initiation of cell death when cells are damaged beyond repair. To identify novel players acting in stress response pathways, we conducted a cell culture RNA interference (RNAi) screen using caffeine as a xenobiotic stress-inducing agent, as this compound is a well-established inducer of detoxification response pathways. Specifically, we examined how caffeine affects cell survival when Drosophila kinases and phosphatases were depleted via RNAi. Using this approach, we identified and validated 10 kinases and 4 phosphatases that are essential for cell survival under caffeine-induced stress both in cell culture and living flies. Remarkably, our screen yielded an enrichment of Hippo pathway components, indicating that this pathway regulates cellular stress responses. Indeed, we show that the Hippo pathway acts as a potent repressor of stress-induced cell death. Further, we demonstrate that Hippo activation is necessary to inhibit a pro-apoptotic program triggered by the interaction of the transcriptional co-activator Yki with the transcription factor p53 in response to a range of stress stimuli. Our in vitro and in vivo loss-of-function data therefore implicate Hippo signaling in the transduction of cellular survival signals in response to chemical stress.
Similar content being viewed by others

Main
Throughout their lives, organisms are exposed to a wide range of harmful substances. These compounds, often referred to as xenobiotics, may enter the body through direct contact, inhalation or ingestion, and can originate from many sources including pharmaceuticals, pesticides, plant toxins and pollutants. The ubiquitous and varied nature of xenobiotic pressure is reflected in the complexity of conserved cellular defense mechanisms that respond to these chemical compounds, and metazoans have thus developed a range of pathways that activate survival responses or trigger programmed cell death to eliminate damaged cells.1 Xenobiotic pathways control the expression and activation of heat shock proteins, oxidative stress components, DNA repair enzymes, hypoxia response factors and several groups of xenobiotic-metabolizing enzymes. In eukaryotes, environmental challenges that activate stress response programs are relayed through a complex signaling cascade. Some stress pathways respond to very specific compounds, whereas others, such as those depending on Mitogen-activated protein kinases, propagate stress signals from a broad spectrum of stimuli.1, 2, 3 Despite the importance that stress response pathways play in pharmacology and toxicology, we still have a relatively poor understanding of the complex networks that launch these cellular defense systems.
We performed a quantitative cell-culture RNA interference (RNAi) assay of Drosophila kinases and phosphatases to identify key regulators of stress response pathways that react to chemical insults. Caffeine is one of the most highly consumed drugs in the world, with well-established pharmacological and toxicological properties, making it an ideal compound for examining cellular stress pathways.4
Caffeine exhibits a broad range of toxic effects in both organisms and cell culture systems, including inhibitory effects on protein and DNA synthesis, disruption of DNA repair mechanisms and induction of apoptosis. Caffeine is a xanthine alkaloid compound that induces a strong detoxification response in flies,5 and this response is—at least in part—mediated by the CncC/Keap1 pathway.5, 6 In addition, caffeine acts as an antagonist of adenosine receptors in the brain, resulting in increased dopamine activity.7
In our screen of the Drosophila kinases and phosphatases, we identified essential modifiers of the survival response to caffeine-induced stress, and remarkably, we noticed these modifiers were 10.7-fold enriched for core components of the Hippo pathway. The Hippo pathway is best known for its role in regulating cell growth, proliferation and survival,8 but in this study, we examined whether Hippo signaling modulates cellular stress responses.
In Drosophila, the core components of the Hippo pathway are the Ste20-like kinase Hippo (Hpo) and the nuclear Dbf2-related kinase Warts (Wts). Upon upstream signaling, Hpo interacts with the scaffold protein Salvador (Sav) to phosphorylate and activate Wts and its cofactor 'Mob as tumor suppressor' (Mats). Activated Wts phosphorylates the transcriptional co-activator Yorkie (Yki). Phosphorylation of Yki results in its retention in the cytoplasm, thus blocking its transcriptional activity.9 When Hippo signaling is inactive, Yki localizes to the nucleus where it forms complexes with transcription factors such as Scalloped,10, 11, 12 Homothorax13 or Mad14 to activate the expression of target genes that promote cell growth, survival and proliferation.15 Yki therefore acts as a growth promoter, whereas Hpo, Wts, Mats and Sav are considered tumor suppressors.
The Hippo core cassette is either activated by signals from the Expanded/Merlin/Kibra (Ex/Mer/Kibra) protein complex16, 17, 18, 19 or from the large atypical cadherin Fat (Ft).20 Ex relays cell surface signals from the trans-membrane polarity protein Crumbs,21, 22, 23 whereas Mer is thought to promote Wts plasma membrane recruitment and activation.24 The Ft atypical cadherin and its ligand Dachsous (Ds) represent another upstream influence on Hippo signaling. Upon binding to Ds and subsequent phosphorylation by the kinase Disc Overgrown (Dco), Ft is thought to antagonize Wts degradation via the myosin Dachs.25, 26 Finally, the actin cytoskeleton has been reported to promote Yki activity (or that of its mammalian counterpart YAP – Yes-activated protein), representing a possible mechanical input into Hippo signaling.27, 28, 29, 30 Although some key molecular mechanisms that regulate Hippo signaling have been identified, many aspects of how these are modulated under physiological or stress conditions remain poorly explored.
In this report, we describe a cell culture-based RNAi screen that identifies the Hippo pathway as a key modulator of cell survival and tissue homeostasis in response to chemical insults. We show that upon stress stimulation, Yki interacts with the stress-responsive transcription factor p53,31, 32, 33 and that the Hippo kinase cascade restrains Yki/p53 activity. Loss of Hippo pathway components leads to dramatic p53-dependent apoptosis and tissue ablation in response to xenobiotic challenge. We suggest that Hippo signaling is required for maintaining tissue homeostasis and promoting cell survival in response to xenobiotics and other forms of stress.
Results
An in vitro assay for cellular modulators of caffeine-mediated stress
To identify novel components acting in stress response pathways, we developed an in vitro assay for measuring cell survival in response to chemical insults. Specifically, we used increasing concentrations of caffeine as a source of xenobiotic stress to challenge Drosophila S2 cells (Schneider 2, derived from embryonic hemocytes). This allowed us to define caffeine levels that trigger a twofold increase in cell death after 24 h of stimulation (Figures 1a and b), which we monitored through a luminescence-based viability assay that detects ATP concentrations (see materials and methods).

Kinome and Phosphatome dsRNA screen for modulators of caffeine-induced cellular stress. (a) Percent viability measured as ATP concentration in Drosophila S2 cells treated with different concentrations of caffeine for ~24 h. (b) Caspase activity assay in S2 cells stimulated for 24 h with 24 or 64 mM caffeine. (c) Flow chart of the RNAi screen. The effect of caffeine on cells is displayed as the ATP/death index. (d) Cells treated with a dsRNA against the anti-apoptotic gene dIAP1 showed a very high ATP/death index relative to control wells treated with a non-silencing dsRNA (directed at GFP). Cells depleted of pro-apoptotic factors, here the Drosophila caspases Ice and Dronc, showed death/ATP indices lower than cells treated with control dsRNA (GFP). (e) Representative plate from the screen. Green indicates genes with ATP/death index >1, while red stands for a death index <1. (f) Distribution of the median dsRNA ATP/death index per gene from a representative plate from the screen. The median ATP/death index for each dsRNA treatment was sorted in descending order. Ninety-nine and 95% confidence intervals are indicated for enhancer and suppressor of cell stress/death. Genes indicated in the diagram represent hits that are statistically significant. (g) Statistical analysis of caffeine-induced cell death. Z-score values >1.96 and z-score <−1.96 represent 95% confidence range
We then tested whether this assay could be adapted to a multi-well format in order to conduct a screen for modulators of caffeine-induced cell death. As positive controls, we depleted the Drosophila effector caspase drICE and the Nedd2-like caspase (Dronc), which should result in reduced levels of cell death. We assayed the effect of an individual dsRNA in the presence and absence of caffeine (each in duplicate), and calculated the death index for each dsRNA as the ratio of cell viability of untreated versus caffeine-treated S2 cells. As expected, we found that the RNAi-mediated knockdown of drICE and Dronc protected cells against caffeine-induced cell death, but exhibited no apparent effect on the viability of untreated controls (Figure 1d). Conversely, knocking down Drosophila inhibitor of apoptosis (DIAP1) as a negative control increased caffeine-dependent cell death sixfold compared with controls (Figure 1d). From these observations, we concluded that our plate-based RNAi approach was sufficient to identify modifiers of caffeine-induced cell death.
Primary RNAi screen in Drosophila S2 cells
To identify novel signaling components of cellular stress responses, we used dsRNA libraries targeting Drosophila kinases and phosphatases. We screened 524 dsRNA (Supplementary Table S1) in S2 cells in quadruplicate in the presence or absence of caffeine for either 3 or 24 h. We calculated caffeine-induced ATP decrease as a ratio of the mean ATP levels without caffeine treatment to the mean ATP concentration of cells treated with caffeine, which we refer to here as the ATP/death index (Figure 1c). We then plotted the median ATP/death index for each dsRNA relative to a non-silencing dsRNA control (directed against GFP) and determined confidence intervals for enhancers and suppressors of caffeine-induced changes in ATP. Thus, we expected that, under caffeine stress, cells depleted of pro-survival gene products would exhibit higher ATP/death indices, whereas depletion of pro-death gene products would exhibit reduced ATP/death indices relative to controls. We calculated z-scores for all dsRNAs to establish thresholds of significance and to enable plate-to-plate comparisons.
Figures 1e and f show a representative 96-well plate from the screen, where we identified PRL-1, the only fly homolog of the human oncogenic protein-tyrosine phosphatases (PRLs), as a pro-death gene with an ATP/death index >95% confidence. Human PRL-3 is a target of the stress response transcription factor p53 and it participates in the p53-dependent response to genotoxic stress.34 PRL-1 was previously reported to inhibit growth and proliferation and to act as a negative regulator of the oncogenic and anti-apoptotic 'C-terminal Src kinase' (Csk) in Drosophila35 as well as in mammals.36 Consistent with this, we identified Csk as a pro-survival gene in our RNAi screen, also with >95% confidence. Taken together, our strategy was suitable for identifying both pro-survival and pro-death genes, although we found that our final list of significant hits is enriched for pro-survival genes (Figure 2).

Whole Kinome and Phosphatome screen for modulators of cell death induced by chemical stress. Quantification of relative ATP content/viability in S2 cells treated with 522 individual dsRNAs and exposed to caffeine for 3 (a) or 24 h (b). The relative ATP/death index z-score was calculated for each dsRNA. The green dashed line represents z-score values >+1.96 (95% confidence) and a z-score>+ 2.58 (99% confidence) while the red dashed line represents a z-score <−1.96 (95% confidence) and a z-score<−2.58 (99% confidence). (c) Heat map of the z-score values for the S2 cells depleted of kinases or phosphatases and stimulated with caffeine for 3 and 24 h
In total, our RNAi screen yielded 54 genes with z-scores that had >95 or >99% confidence intervals (Figure 2). Of the 524 dsRNAs we analyzed (Supplementary Table S1), we identified 28 genes for each time point (3 and 24 h) that acted as pro-survival modifiers in the RNAi screen, resulting in a total of 51 unique genes (5 genes were found at both time points: CycA, CycT, sds22, warts and CG9324). In contrast, the screen yielded only two and one pro-death genes at 3 and 24 h, respectively, resulting in a total of three unique genes.
Secondary RNAi screen in Drosophila
To validate our cell-based screen, we performed an in vivo RNAi counter-screen for each of the 54 hits. For this, we crossed fly stocks obtained from the Vienna Drosophila Resource Centre (VDRC) library to a total of five different GAL4 drivers, which allowed us to produce dsRNA in specific tissues for each of the 54 genes. We triggered RNAi in the gut (Mex-Gal4), the Malpighian tubules (C724-Gal4) and in the fat body (Cg-Gal4), which represent key tissues required for xenobiotic metabolism, and should thus contribute to cellular survival in the presence of xenobiotic compounds such as caffeine. This approach allowed us to quantify lethality rates by scoring for adults that survived from a defined population of eggs (Figure 3a). In addition, we assessed the effects of RNAi depletion in eye imaginal discs using an eyeless Gal4 driver (ey-Gal4). The proliferation and apoptosis patterns for the Drosophila compound eye are well defined. In addition, the adult eye is dispensable for survival under laboratory conditions, making it an ideal tissue for identifying loss-of-function phenotypes that result from increased cell death (Figure 3b). Finally, we also examined the effects of producing dsRNA ubiquitously for each of the 54 candidate genes, by way of the actin-Gal4 driver. This served to assess whether the RNAi transgene elicited expected phenotypes, and to establish a reference that could be compared with the tissue-specific knockdowns.

In vivo validation of primary screen hits based on tissue-specific RNAi. (a) Survival rates for eclosed adults where RNAi was triggered in the Malpighian tubules (C724), the fat body (Cg) or the midgut (Mex). Survival rates for each Gal4 × UAS-RNAi cross are shown as the percentage of embryos that developed to adulthood in a given experiment. n=150 eggs per each genotype and condition. Black bars: regular food, white bars: 4 mM caffeine, gray bars: 1 mM phenobarbital. Error bars represent standard deviation; P-values (not shown) are based on Student’s t-test and are all P<0.01 for the affected condition. (b) Eye-specific phenotypes when RNAi is triggered in the developing eye, tested on regular, caffeine or phenobarbital media
For this secondary screen, we placed 3 × 50 eggs for each cross (5 Gal4-drivers × 54 RNAi lines=270 crosses) on fly media that contained either 0 or 4 mM caffeine. In addition, we tested media containing another compound, phenobarbital (PB), to examine whether responses were specific to caffeine or of a more general nature. Like caffeine, PB is a potent inducer of detoxification responses in the fly,37 but its biological activity is based on a different mechanism, because PB acts by stimulating GABA receptors. We then scored for obvious phenotypes (morphological changes and lethality) on either media type. Of the 54 RNAi lines we tested, 34 crosses displayed lethality when RNAi was triggered ubiquitously, and another 2 crosses (otk and sds22, see Table 1) exhibited lethality only when reared on caffeine- or PB-containing media. When we tested the same 54 RNAi lines with tissue-specific Gal4 drivers, we found six lines that caused lethality regardless of whether PB or caffeine was present, while a total of 14 lines exhibited drug-specific phenotypes ranging from lethality to eye phenotypes (Table 1). In all 14 cases, a caffeine-induced phenotype was mirrored by a corresponding PB-dependent defect, although differences in the class or strength of a given phenotype varied with the drug being used. Using two structurally different drugs not only confirmed our findings, but also strongly suggested that our caffeine-based results are representative of a general xenobiotic response.
Our secondary screen validated a total of nine pro-survival genes associated with tissues that metabolize xenobiotics: three genes with specific roles in the midgut (dnk, PhKγ, sds22), two in the fat body (CycT, Pp2A-29B), three in the Malpighian tubules (CanA1, Csk, mRNA-cap), and one that appears to be important in both, the Malpighian tubules and the midgut (Tie) (Figure 3a and Table 1). When we assessed the effects of RNAi depletion of candidate genes in the eye tissue, we identified eight genes with drug-dependent phenotypes, three of which were also identified in the tissues with xenobiotic functions (dnk, mRNA-cap, sds22), whereas the remaining five appeared to be unique to the eye (Adk2, dco, otk, polo, warts) (Figure 2b and Table 1).
Taken together, the secondary in vivo RNAi screen validated 14 genes out the 54 hits from the primary in vitro RNAi screen and identified tissue-specific roles for each gene during drug-induced stress. Our results also indicate that these genes might act in a general stress response pathway, as two independent drugs caused comparable results.
The Hippo pathway promotes cell survival in response to chemical stress
To investigate potential relationships among the 14 genes that remained after our secondary screen, we mapped the genetic and physical interactions into a representative network by utilizing databases of reported and predicted interactions38, 39 (Figure 4a). Remarkably, the majority of our strongest screen hits were associated with the core kinases of the Hippo pathway.8 We found that of the 524 kinases and phospatases in our library, 14 genes were annotated as part of the Hippo pathway interactome (2.66% of the library). We initially identified 54 candidate hits out of the 524 original genes in the primary screen, and 6 out of the 54 hits were reported as part of the Hippo interactome (11.1%). In our secondary screen, we confirmed 14 out of the 54 initial hits and 4 out of these 14 hits have been reported as Hippo pathway modulators (28.6%). Thus, after two rounds of screening, Hippo pathway modulators were enriched 10.7-fold.

Hippo pathway players are negative regulators of caffeine-induced cell death. (a) Partial genetic and physical interaction network of modifiers of xenobiotic stress (see materials and methods). Black circles represent hits that were identified in the primary screen and confirmed in the secondary screen. The grey circle represents a hit in the primary screen that was not confirmed in the secondary screen. White circles represent components of the Hippo pathway that were not identified in our screen. (b) Eye-specific RNAi phenotypes of hippo (hpo) and yorkie (yki). (c) Caffeine induces increased level of phosphorylated form of Hpo kinase in control animals (ey>w1118). Hpo and P-Hpo blots represents two different gel loads of the same protein extracts to eliminate the possibility of incomplete stripping. (d) Genetic interactions between Hippo pathway players. Gene names in red indicate RNAi transgene, while gene names in green indicate transgenes carrying a corresponding cDNA. From left to right, we show eye phenotypes in flies of the following genotypes: [ey>UAS-dco-RNAi], [ey>UAS-wts-RNAi], [ey>UAS-dco-RNAi; UAS-wts-RNAi], [ey>UAS-hpo-RNAi; ey>UAS-dco-RNAi], [ey>UAS-hpo-cDNA], [ey>UAS-hpo-cDNA; UAS-dco-RNAi], [ey>UAS-yki-RNAi; UAS-dco-RNAi], [ey>UAS-yki-cDNA], [ey>UAS-yki-cDNA; UAS-dco-RNAi]. All crosses were performed with flies raised on media containing either 0 mM (top panels) or 4 mM caffeine (bottom panels)
In particular, we identified Warts (Wts), a Ser/Thr kinase acting directly downstream of Hpo, and Discs Overgrown (Dco), also a Ser/Thr kinase, which is known to promote Wts activity downstream of Ft.9 We also identified the known Hippo pathway modulators Pp2A 40 and Csk.41 Although the Hippo signaling pathway is best characterized for controlling tissue size, some work in Drosophila has linked the pathway to genotoxic stress42 and alcohol-induced stress.43 These studies, however, did not propose a molecular mechanism that could explain the role of Hippo as a modulator of stress response pathways.
Our screen revealed that the depletion of wts and dco affected cell survival in vitro (Figure 2), and caused almost complete ablation of the adult eye upon caffeine treatment in vivo (Figure 3b). We therefore examined whether the depletion of Hpo kinase would result in a comparable phenotype, to establish whether or not the caffeine-dependent wts- and dco-RNAi phenotypes were related to their reported functions within the Hippo pathway.9 To this end, we depleted hpo specifically in the developing eye (ey>hpo-RNAi) of Drosophila larvae reared on regular or caffeine-supplemented food (Figure 4b). As expected, we found that interfering with hpo in eye imaginal discs resulted in overgrown eye tissue on regular food. Strikingly, when animals of the same genotype were grown on caffeine-containing food, adult eye formation was completely abolished (Figure 4b). Thus, the core kinases of the Hippo pathway, Hpo and Wts, are required to maintain tissue integrity in response to caffeine-induced stress.
To confirm our data, we analyzed the effect of caffeine on hpo, wts and dco mutant cells generated using the FLP-FRT system. Mutant tissues were generated in the eye using Flipase expression controlled by the eyeless promoter (eyFLP) and marked by the loss of eye pigmentation in the adult eye. Clones of cells homozygous for the dco3 allele were eliminated in eyes of flies reared on caffeine food (area highlighted by dashed line), whereas mutant cell clones survived when animals were raised on regular food (Supplementary Figure S3A). Similarly, cell clones homozygous for wts or hpo were either sparse or absent in eyes of flies grown on caffeine food, in contrast to the strong overgrowth of these clones in flies fed on regular medium (Supplementary Figure S3A). Our data gathered from the dco, wts and hpo mutant clones therefore were consistent with our results obtained from RNAi-depleted tissues, with both approaches revealing strong caffeine-dependent phenotypes.
As Dco is known to promote Wts activity downstream of Ft,44 we tested the effect of ft knock down in the developing eye of animals in the presence or absence of caffeine. This resulted in a slightly rough eye phenotype in caffeine-treated animals (data not shown). It was possible, however, that the ft-RNAi lines were not very effective inhibitors of ft function, prompting us to further examine the role of Ft using the FLP-FRT system to generate clones of cells homozygous for a ft mutant allele (fatfd) in developing eye. We observed that the majority of ft mutant clones were absent in adult mosaic eyes of animals raised on caffeine media (area highlighted by dashed line in Supplementary Figure S1C), whereas ft mutant clones were abundant and enlarged in animals raised on regular food, as expected (Supplementary Figure S1C). Thus, the role of Dco in promoting cell survival under stress is likely dependent on the activation of Ft to modulate the Hippo pathway stress response.
Hippo kinase is activated by caffeine-induced stress
We examined whether caffeine could promote activation of Hippo kinase in vivo. We monitored the phosphorylation status of residue T195 in the kinase domain activation loop, which is required for Hpo activity.42 We analyzed the levels of phosphorylated Hpo kinase by western blotting, and compared control animals (third instar (L3) larvae of ey>w1118) fed with 0 or 4 mM caffeine. In agreement with the notion that many pro-apoptotic stimuli can promote Hpo or mammalian MST1/2 activation,42, 45, 46 we found markedly higher levels of phospho-Hpo in extracts of eye imaginal disc isolated from larvae reared on a caffeine diet compared with controls (Figure 4c), indicating that caffeine promotes the activation (= phosphorylation) of Hpo kinase.
Once we had established that the Hippo pathway was involved in the cellular response to caffeine, we performed genetic interaction experiments to examine the relationships between dco and the principal kinases acting in the Hippo cassette. When we co-depleted hpo and dco in the developing eye of larvae raised on normal food, we observed a tissue overgrowth phenotype, consistent with the downregulation of Hippo signaling. When these ey>hpo/dco-RNAi flies were raised instead on caffeine media, the eye tissue was completely ablated, similar to what we observed when dco or hpo were knocked down individually (also on caffeine media). However, when we overexpressed a hpo cDNA in an ey>dco-RNAi background, we observed a dramatic rescue of eye development in caffeine-fed flies compared with dco-RNAi alone (Figure 4d), suggesting that Dco acts through Hippo or in a parallel pathway to respond to xenobiotic stress. This is consistent with the notion that Ft/Dco signaling can promote Wts activity by increasing its stability.
Hippo signaling inactivates the transcriptional co-activator Yorkie (Yki) via phosphorylation at three sites.47, 48 We therefore tested the involvement of Yki in caffeine-induced toxicity. Consistent with its role as a growth-promoting factor, eye-specific loss of yki function via RNAi resulted in a small eye phenotype on regular food and in the presence of caffeine (Figure 4b). As depletion of hpo kinase results in the hyperactivation of Yki, we hypothesized that the abrogated eye development observed in ey>hpo-RNAi or ey>wts-RNAi flies grown on caffeine was a consequence of increased Yki activity. As previously reported,49 eye-specific overexpression of yki causes overgrowth of eye tissue (Figure 4d). When flies of the same genotype were reared on caffeine food, they displayed abnormally round eyes, which were also rough and appeared to contain less pigment, suggesting that caffeine exposure reduced the eye overgrowth phenotype because of increased apoptosis (Figure 4d). This was a surprising observation, as Yki is known to promote the expression of DIAP1, the principal Drosophila caspase inhibitor, which is expected to render cells more resistant to apoptotic signals.
When a yki cDNA was overexpressed in an ey>dco-RNAi background, the overgrowth phenotype was slightly more pronounced compared to yki overexpression alone on regular food, however, eye development was completely abolished when ey>dco-RNAi; UAS-yki-cDNA flies were reared on caffeine. These data may suggest that when Yki alone is overexpressed, the Hpo kinase cascade is still intact and therefore able to rescue some cells by silencing Yki under caffeine treatment. Thus, when dco is inactivated in the presence of overexpressed yki, the core kinase cascade would no longer be able to restrain Yki activity, thereby uncovering a pro-apoptotic function for Yki under stress conditions. In Drosophila, the best-characterized transcriptional partner of Yki is Scalloped (Sd), a TEAD/TEF protein family member.10, 11 We therefore wanted to test whether Sd functions in conjunction with Yki in mediating the pro-apoptotic response to caffeine. Depletion of sd via RNAi caused a small eye phenotype as previously reported12 in flies raised on regular media; however this small eye phenotype was not altered on caffeine media. We then analyzed the effect of sd depletion in an ey>dco-RNAi background, which showed that while flies reared on regular food developed relatively normal eyes (resembling the ey>UAS-dco-RNAi phenotype), flies grown on caffeine media failed to develop eyes, similar to what we observed when dco alone was depleted (Supplementary Figure S2). When ey>wts/sd-RNAi flies were reared instead on caffeine media, eye tissues were completely ablated, again similar to what we observed when wts alone was knocked down in flies reared on caffeine media. Finally, we depleted sd and hpo together in the developing eye of Drosophila larvae reared on regular or caffeine food (Supplementary Figure S2). As previously observed,12 we found that the co-depletion of sd and hpo in eye imaginal discs resulted in overgrown eye tissue on regular food. Strikingly, when animals of the same genotype were reared on caffeine-containing food, adult eye formation was completely abolished, as is the case when hpo alone is depleted (Supplementary Figure S2). Taken together, these data strongly suggest that Sd is not required for the Yki-mediated pro-apoptotic response to caffeine.
Caffeine induces apoptosis by activation of p53
Herein and in previous work, we have established that caffeine-induced stress ultimately leads to apoptosis in a cell culture system (Figure 1b) and in live animals.50 To test whether the loss of eye tissue in caffeine-reared ey>dco-RNAi animals is a consequence of increased apoptosis, we ectopically expressed the anti-apoptotic p35 gene in this background. p35 expression resulted in a complete rescue of the caffeine-dependent disruption of eye development, demonstrating that excessive apoptosis was responsible for the lack of eye tissue (Figure 5a). This was also the case when we expressed p35 in an ey>wts-RNAi background (Supplementary Figure S1B). We did not observe any rescue in control flies that expressed EGFP instead of p35 as a second transgene in ey>dco-RNAi flies. Similar to the p35 rescue, expression of a dco-cDNA in ey>dco-RNAi flies also rescued the eye phenotype in the presence of caffeine (Figure 5a).

dco protects cells from p53-induced apoptosis. (a) Expression of the anti-apoptotic gene p35 from baculovirus rescues the ablated eye phenotype associated with eye-specific knockdown of dco animals when raised on caffeine food. Overexpression of the Egfp transgene in a dco-depleted background serves as a negative control. (b) Eye-specific co-depletion of the pro-apoptotic transcription factor p53 in ey>dco-RNAi and ey-wts-RNAi backgrounds rescues caffeine-dependent ablation of eye phenotypes. (c) Relative expression of the pro-apoptotic gene reaper in larval heads of ey>dco-RNAi and ey>wts-RNAi. Error bars represent standard deviation. Flies were reared on normal medium (=no caffeine). P-values are not shown, but rpr expression levels in dco and wts-RNAi animals were significantly different from the control (P<0.001; Student’s t-test, n=4 biological replicates for each genotype)
Multiple reports suggest that, under some forms of stress, the mammalian Yki ortholog, Yes-associated protein (Yap) switches from an anti-apoptotic to a pro-apoptotic function.51, 52, 53, 54, 55, 56, 57 In many of these cases, this pro-apoptotic function has been linked to YAP’s ability to bind to p73 and promote p73-dependent gene expression. As Drosophila p53 is most closely related to mammalian p73, we tested the involvement of Drosophila p53 in caffeine-induced toxicity. We observed that co-depleting p53 with either ey-wts- or ey-dco-RNAi completely rescued the caffeine-dependent eye ablation phenotype (Figure 5b), suggesting that Hippo signaling normally suppresses p53-dependent apoptosis in response to caffeine.
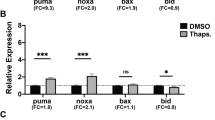
As several mammalian reports indicated a stress-dependent interaction between YAP and p73, we tested whether Drosophila p53 was associated with Yki under various stress conditions. We performed co-IP experiments in S2 cells between Yki and p53 upon caffeine stimulus. We observed that p53 interacted robustly with Yki when cells were treated with caffeine, whereas the interaction was not detected in untreated cells (Figure 6a). We then examined whether this interaction would occur under a range of stress stimuli. Interestingly, we observed a Yki-p53 interaction upon treatment with cadmium chloride and sodium arsenate, which are both known to generate oxidative stress in cells, as well as UV irradiation and heat shock treatment (Figures 6b and d). Thus, Yki may function as part of a pro-apoptotic complex with p53 that forms upon treatment with a range of stimuli. One of the best-characterized transcriptional targets of p53 encodes the pro-apoptotic protein Reaper (rpr), which promotes apoptosis by antagonizing DIAP1 function.33, 58 This prompted us to measure the expression of rpr transcripts in dco- or wts-depleted larval heads using ey-Gal4 as a driver, as well as controls (Figure 5c). Interfering in this manner with Wts or Dco function led to an increase in rpr expression, suggesting that the Hippo pathway normally functions to restrict p53 activity in larval eyes. Taken together, our results indicate that stress stimuli such as caffeine promote cell death via the formation of a p53/Yki complex. The Hippo core kinase cascade normally dampens this pro-apoptotic response, thus preventing excess apoptosis triggered by stress-induced Yki/p53 activity (Figure 7).

Hippo pathway components interact with p53. (a) Co-immunoprecipitation (co-IP) of FLAG-tagged Yki and HA-tagged p53 in cells treated with 0 or 4 mM caffeine. Upper panels: input, lower panel: FLAG-IP. Arrow: HA-p53. (b) co-IP of FLAG-tagged Yki and HA-tagged p53 in cells treated with ROS-inducing CdCl (lane 1), mock-treated (control, lane 2) and ROS-inducing NaAsO2 (lane 3). Upper panel=input, lower panel=FLAG-IP. Asterisk=unspecific band, arrows: FLAG-Yki. (c) co-IP of Flag-tagged Yki and HA-tagged p53 in UV-treated and non-treated cells. Asterisk: unspecific band, arrows: HA-p53. (d) co-IP of Flag-tagged Yki and HA-tagged p53 in heat-shock treated (37 °C) and non-treated (25 and 4 °C) cells. Asterisk: unspecific band

Model for role of Hippo pathway in stress responses. Xenobiotic and other forms of stress induce an apoptotic stimulus driven by Yki, which interacts with the stress response transcription factor p53. The strong apoptotic response needs to be modulated in order to prevent cell death and tissue degeneration. The Hippo pathway is activated in the presence of cellular stress and acts as negative regulator of Yki/p53-induced cell death
Discussion
Our screen identified the kinases Wts and Dco, both members of the highly conserved Hippo pathway (MST1/2 in humans). The dramatic tissue overgrowth phenotypes associated with loss-of-function mutations in any of these pathway components demonstrates their potent tumor suppressor activity.8 Indeed, we observed this striking overgrowth phenotype in eye tissues when we triggered RNAi against dco and wts (Figure 3b). Remarkably, when ey>dco and ey>wts-RNAi animals were instead reared on caffeine-containing media, the eye tissue was perfectly ablated. These findings corroborate the idea that the Hippo pathway plays an important role in mediating responses to xenobiotic stress. Activation of Hpo kinase occurs through phosphorylation of its kinase domain (on T195),42 and consistent with this, we found increased levels of phosphorylated Hpo in flies fed with caffeine, suggesting that drug exposure promotes Hpo activation.
As the primary function of the Hippo pathway is associated with growth control, we tested whether caffeine-induced stress affects other growth control pathways during eye development. Importantly, when we manipulated the levels of Insulin-like receptor (InR) 62 (Supplementary Figure S1A), a well-known regulator of growth, we observed no caffeine-dependent phenotypes, indicating that cellular growth pathways per se are not involved in the response to chemical stress.
The key target of the Hippo kinase cascade that effectuates growth control is Yki, a transcriptional co-activator.63 Yki has been classified as an oncogene, as it promotes proliferation and growth under normal conditions, and thus inhibits apoptosis.15 Consequently, inactivation of the core kinase cascade causes tissue overgrowth because of Yki de-repression. If the caffeine-dependent ablation of eye tissue we observed in hpo- or wts-RNAi animals were indeed a consequence of Yki activity, then one would expect Yki overexpression (ey>yki-cDNA) to produce a comparable eye phenotype. However, although the level of overgrowth caused by yki overexpression is reduced in the presence of caffeine, the reduction in eye size is not as dramatic as that observed in ey>dco-RNAi or ey>hpo-RNAi animals (Figure 4d). A plausible explanation is that Hpo signaling is still operational in ey>yki-cDNA flies and therefore can still partially repress overexpressed Yki, therefore dampening the effect of caffeine treatment on cell viability. Consistent with this idea, we find that Yki overexpression in an ey>dco-RNAi background resulted in complete eye ablation on caffeine (Figure 4d). Thus, we hypothesize that Yki may act as either a pro-growth or pro-apoptotic factor dependent on cellular context.
The mammalian homolog of Yki, YAP, is capable of interacting with a range of transcription factors and target genes, depending on the cellular context. For instance, YAP interacts with TEAD transcription factors (Transcriptional enhancer activator domain—Scalloped in flies) to stimulate the expression of growth-promoting and anti-apoptotic targets, thereby driving cell proliferation and survival.10, 11, 12 In contrast, when cells are chemically stressed via cisplatin treatment, YAP binds to and stabilizes the p53 family member p73, thus promoting pro-apoptotic gene expression.52, 53, 54 Similar to the YAP/TEAD pro-growth/survival function, the pro-apoptotic role of YAP/p73 is suppressed by the core Hippo signaling cascade.51, 56
Drosophila has a single member of the p53 family, p53,31, 33 which regulates cell death and differentiation pathways.32 Drosophila p53 is most closely related to mammalian p73. We observed that co-depleting p53 in combination with either wts- or dco-RNAi completely rescued the caffeine-dependent eye ablation phenotype (Figure 5b), whereas sd co-depletion with wts, hpo or dco had no effect (Supplementary Figure S2). This suggests that Hippo signaling normally suppresses p53-dependent apoptosis in response to caffeine. On the basis of the YAP/p73 interaction, we hypothesized that Yki might bind to p53 and drive the expression of pro-apoptotic genes. Indeed, in the presence of different cellular stress stimuli, Yki co-immunoprecipitates with p53 in Drosophila S2 cells (Figure 6). Moreover, wts or dco depletion increased reaper mRNA levels in animals reared on normal food (Figure 5c), consistent with the finding that reaper is a p53 target gene. We therefore reason that the activation of the Hippo pathway is required to restrain p53/Yki-induced apoptosis when cells are exposed to chemical stress (Figure 7), thereby preventing excess cell death and potentially allowing for subsequent tissue repair.
Interestingly, we have previously shown that upon γ-irradiation, which also activates Hpo, loss of the core kinase cascade protects rather than sensitizes wing disc cells against p53-dependent apoptosis.42 This suggests that the context-dependent role of YAP in preventing or promoting stress-induced apoptosis is conserved in Drosophila Yki. The exact molecular nature of the context-dependent cue(s) that determine this 'apoptotic switch'57 remain to be established, but are likely to involve posttranslational modification of Yki and/or p53, as well as the activation status of other signaling cascades, such as the JNK pathway 64 or c-Abl,65 both of which have been shown to drive YAP-dependent apoptosis. The conserved nature of this dual behavior of Yki in damage response may reflect its proposed ancient function as a mediator of tissue damage repair.66 This decision-making process may lie at the heart of the choice each damaged cell must make between survival and repair or undergoing apoptosis in case of irreparable damage. Given the mounting evidence that YAP is activated and promotes oncogenic transformation in a broad spectrum of tumors,67 the elucidation of this switch mechanism may offer therapeutic opportunities by tipping the balance between life and death of tumor cells.
Materials and Methods
Cell culture
Drosophila S2 cells were cultured at 25 °C in HyQ TNM-FH medium (GE Healthcare Life Sciences, HyClone Laboratories, Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 50 g/ml of penicillin and 5 g/ml streptomycin (GIBCO, Life Technologies Inc., Burlington, ON, Canada). Serum-free S2 cells were grown in SFX-INSECT medium (Hyclone) supplemented with 50 g/ml of penicillin and 5 g/ml streptomycin (GIBCO).
Caspase activation assay
S2 cells (5 × 105 cells/well in 6-well plates) were treated with 0, 24 or 64 mM caffeine (Sigma-Aldrich Canada Co., Oakville, ON, Canada) for 24 h to induce caspase activation. Each concentration was tested in triplicate in three independent experiments. Caspase-3/7 like activation was quantified using a fluorescence-labeled DEVD peptide substrate assay (Sigma-Aldrich Canada Co.). Fold caspase-3/7 like activity was calculated as the ratio between the mean caspase-3/7 activity of cells treated with 24 mM (or 64 mM) caffeine and the cells treated with 0 mM caffeine.
RNA interference
The kinome and phosphatome library used in this screen was extracted from a genome library described previously.59 Drosophila S2 cells (105 cells/well in 96 wells) were incubated in serum-free culture media with 10 g/ml dsRNA at 25 °C for 3 days. Each source plate was transfected in quadruplicate. Subsequently, cells in two plates were treated with 24 mM caffeine for 24 h. The screen was repeated in the presence of caffeine for 3 h. Cell viability was assessed by measuring cellular ATP levels (CellTiter-Glo luminescent cell viability assay; Promega Corporation, Madison, WI, USA). ATP concentrations were normalized to the mean ATP concentration of the mock non-silencing dsRNA control (directed against GFP) in the same plate. ATP concentrations were averaged for replicate plates. The mean ATP values with and without treatment were used to determine the death/ATP index for each dsRNA. The death/ATP indices were defined as the ratios of ATP values from cells without caffeine treatment to the ATP values from cells stimulated with caffeine. The analysis assigned to the mock non-silencing dsRNA (GFP) a death/ATP index value of one. We applied z-score analysis to normalize death indices across the entire screen. The z-scores were calculated by subtracting the death index value for each dsRNA by the plate median value and dividing by the standard deviation of the plate. The z-scores assume normal distribution and represent the standard deviation of every death index from the plate median for each dsRNA treatment. Z-scores above 2.58 and below −2.58 represent the 99% confidence of interval and z-score above 1.96 and below −1.96 represents the 95% confidence of interval.
Data analysis
The heat map of z-score values for the hits of the screen at 3 or 24 h was created using java tree view (http://jtreeview.sourceforge.net/).60 To visualize interactions between the hits of our screen we probed all hits in the Drosophila interaction database38 and STRING.39
Fly stocks
The UAS-sd-RNAi line was a kind gift of the Simmonds laboratory. All other UAS-RNAi transgenic fly lines were obtained from the VDRC RNAi library. UAS-Hpo and UAS-Yki were kind gifts from Nobert Perrimon laboratory. yw; FRT82B Dco3 was a gift fom Helen McNeill yw; FRT42D, hpo42, 43, 44, 45, 46, 47, 48/Cyo, ey-FLP, Ubi-GFP, [w+] FRT42D, yw;FRT82B,wtsM541/TM6B, yw; FRT40A ft fd, ey-FLP, Ubi-GFP, [w+] FRT40A, ey-FLP, Ubi-GFP, [w+] FRT42D were from Nic Tapon laboratory. ey-FLP, UAS-mCD8::GFP; tubGal80 FRT82B, mex-Gal4, c724- Gal4, act-Gal4, cg-Gal4 and ey-Gal4 were obtained from the Bloomington Drosophila stock center.
Drosophila strains were cultured on standard cornmeal medium (http://flystocks.bio.indiana.edu/Fly_Work/media-recipes/bloomfood.htm) at 25 °C. Flies used in the drug survival assay were cultured on standard cornmeal medium supplemented with 4 mM caffeine or 1 mM PB (both Sigma-Aldrich Canada Co.).
Secondary screen and survival screen
All UAS-RNAi lines were crossed to Mex-Gal4, C724- Gal4, act-Gal4 and Cg-Gal4. Fifty eggs of each genotype were transferred to vials containing standard medium, or standard medium supplemented with either 4 mM caffeine or 1 mM PB. The eggs were left to develop to the adult stage. We counted the number of adults eclosed and calculated the survival rate for each genotype as the percentage of adults eclosed compared with controls. Each genotype was tested in triplicate for a total of 150 eggs. Statistical significance was expressed as P-values of less than 0.01 (**) or 0.05 (*), as calculated by a Student’s t-test (two-tailed).
Secondary screen and eye tissue analysis
All UAS-RNAi lines were crossed to the ey-Gal4 driver on a standard diet, and standard diets supplemented with 4 mM caffeine or 1 mM PB. After 3 days, the parental generation was removed from the vials and eggs were left to develop into adults. Two to 3 days after eclosion, adults were examined visually for alterations in eye morphology. w1118crossed to ey-Gal4 served as a control strain. Each genotype was tested in triplicate.
Preparation of protein extracts
Ten Drosophila L3 larvae were staged at 24 h after the L2-L3 molt. Heads were dissected from staged animals and mixed with 300 μl of cold lysis buffer (Ephrussi–Beadle Ringer's solution with 10 mM EDTA, 10 mM DTT, and Roche Complete Protease Inhibitor, Hoffmann-La Roche Limited, Mississauga, ON, Canada). Then, 150 μl of hot (70 °C) 3 × SDS-PAGE sample buffer containing 10 mM DTT was added to the homogenate and incubated at 100 °C for 10 min. Particulate matter was pelleted at 13 000 r.p.m. for 2 min, and the supernatant transferred to a new tube.
DNA cloning
Drosophila melanogaster total RNA was prepared from pupal tissue using the RNAeasy kit (Qiagen, Toronto, ON, Canada) according to the manufacturers protocol. Yki-transcripts were then reverse-transcribed with superscript III reverse polymerase (Life Technologies Inc.) with primer CACC-Yki-5′, 5′-C ACC ATG TGC GCG TGC CTA ATC GCT AAG-3′, Yki-STOP-3′, 5′-TTA ATT AAT TTT ATA CCA TTC CAA ATC GTC AGG TTT GTG G-3′ and Yki-NoSTOP-3′, 5′-ATT AAT TTT ATA CCA TTC CAA ATC GTC AGG TTT GTG G-3′ for yki cDNA with and without a stop codon, respectively. Purified PCR products were then cloned into the pENTR-D-TOPO vector (Life Technologies Inc.), sequence-verified for the long isoform of Yorkie (Uniprot: Q45VV3-1, CG4005) and subsequently cloned into pActin5C destination vectors bearing various tags (Drosophila Gateway Vector Collection) via the Gateway method (Life technologies Inc.). The cDNA for p53 was obtained from pAWH-p53 (Colombani et al.42) with PCR using primers Dmp53PB-5’, 5′-C ACC ATG AGT CTT CAC AAG TCC GCG TCG-3′ and Dmp53nostop-3′, 5′-TGG CAG CTC GTA GGC ACG TTT CTT AAG-3′.
S2 cell transfection, stress stimulation and immunoprecipitation
Equal amounts of Schneider S2 cells were grown in HyQ TNM-FH medium (HyClone) in 6-well plates to ~90% confluency (1.6–1.8 × 106 cells/well), transfected with 300-400 ng plasmid DNA using Cellfectin II (Invitrogen) following the manufacturer protocol and exposed to various stimuli:
Caffeine stimulation
Cells were treated with 4 mM caffeine (in cell media), control cells were mock-treated with cell media and samples were incubated for 3 h.
UV samples
were exposed to 300 J/m2 of UVC-rays using a Hoefer, Inc. (Holliston, MA, USA) UVC Illuminator after removing and collecting the S2-medium for each well and washing with PBS. Cells were then incubated in their conditioned medium for 15 min (sample medium from each well was removed and kept at 25 °C prior to UV exposure).
ROS-inducing agents
Cells were treated with proteasome inhibitor (MG132) and 100 μM CdCl or 200 μM NaAsO2 (in DMSO), control cells were mock-treated with DMSO and samples were incubated for 2.5 h.
Heatshock
Cells were exposed to 25 °C, 37 °C or 4 °C for 15 min prior to lysis on ice.
After treatment, cells were washed once with PBS and lysed with ice-cold lysis buffer (150 mM NaCl, 50 mM Tris (pH 8), 0.5% NP40, 1 mM EGTA, 0.5 M NaF, 200 mM vanadate, phosphatase inhibitor cocktail 1 (Sigma-Aldrich Canada Co.), and protease inhibitors (Roche) and immunoprecipitated using M2-FLAG-Agarose beads (Sigma-Aldrich Canada Co.) for 3–4 h. FLAG beads were boiled for 5 min at 70 °C in SDS sample buffer and proteins were electrophoresed on SDS-PAGE (NuPage 10% Bis-Tris precast gels).
Immunoblotting
Proteins were electrophoresed at 170 V by SDS-PAGE on precast 4–12% Bis-Tris polyacrylamide gels (Novex, Life Technologies Inc.), and transferred onto nitrocellulose membranes in a Trans-Blot apparatus (Bio-Rad Laboratories (Canada) Ltd., Mississauga, ON, Canada). Membranes were blocked in blocking buffer (Li-COR Biosciences, Lincoln, NE, USA) for 1 h at room temperature and then incubated for 1 h with primary antibody in TBSTw (Tris-buffered saline (150 mM NaCl, 20 mM Tris pH 7.5+0.05% Tween 20). After washing three times for 5 min with TBSTw, membranes were incubated with a secondary antibody for 1 h at room temperature. Finally, membranes were washed as above in Tris-buffered saline plus 0.2% Triton X and immune-complexes detected by enhanced chemiluminescence (ECL; Amersham Biosciences, Baie d'Urfe, QC, Canada) or by Odyssey Infrared Imaging System (LI-COR Biosciences).
Primary antibody dilutions used in immunoblotting experiments were as follows: rat anti-HA (Roche, 1 : 500 to 1 : 5000), rat anti-Hippo,61 rabbit anti-P-Mst1/2 (Cell Signaling Technology, New England Biolabs, Ltd., Whitby, ON, Canada, 1 : 500), rabbit anti-Warts (kind gift from Prof. Ken Irvine, 1 : 500), rabbit anti-DIAP1 (Kind gift from Prof. Hyung Don Ryoo, 1 : 500), rabbit anti-FLAG (Sigma-Aldrich Canada Co., 1 : 1000), mouse anti-atubulin (Sigma-Aldrich Canada Co., 1 : 1000). IRDye 700 and 800 secondary antibodies were used at a 1 : 10 000 dilution. Horseradish peroxidase-linked secondary antibody (Amersham) was used at a 1: 10 000 dilution.
RNA extraction and qPCR
Ten Drosophila L3 larvae were staged at 24 h after the L2-L3 molt. Heads were dissected from these animals and placed in RNAse-free 1.5 ml tubes. RNA was extracted with the RNeasy Plus Kit (Qiagen) according to the manufacturer's instructions. RNA samples (0.5–2 μg per reaction) were reverse-transcribed using ABI High Capacity cDNA Synthesis kit, and the resuling cDNA was used for qPCR analysis (StepOnePlus, Applied Biosystems, Life Technologies Inc.), using KapaSYBR PCR master mix (Kapa Biosystems, Inc., Wilmington, MA, USA) with 5 ng of cDNA template and a primer concentration of 200 nM. Samples were normalized to rpl23 (rpl23-reverse 5′ GTT TGC GCT GCC GAA TAA CCA C3′ and rpl23-forward 5′ GAC AAC ACC GGA GCC AAG AAC C3′) based on the ΔΔCT method. Reaper transcripts were detected using the following oligos: Reaper Left 5′-TCG ATT TCT ACT GCA GTC AAG G3′ and Reaper Right 5′-GAG TAA ACT AAA ATT GGG TGG GTG T3′.
Abbreviations
- Hpo:
-
Hippo
- Wts:
-
Warts
- Dco:
-
Disc overgrown
- Sav:
-
Salvador
- Mats:
-
Mob as tumor suppressor
- Ex:
-
Expanded
- Mer:
-
Merlin
- Ft:
-
Fat
- Yki:
-
Yorkie
- Ds:
-
Dachsous
- Yap:
-
Yes-act protein
- L3:
-
third instar
- co-IP:
-
co-immunoprecipitation
References
Fulda S, Gorman AM, Hori O, Samali A . Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010; 2010: 214074.
Shiloh Y, Ziv Y . The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14: 197–210.
Matsukawa J, Matsuzawa A, Takeda K, Ichijo H . The ASK1-MAP kinase cascades in mammalian stress response. J Biochem 2004; 136: 261–265.
Henderson-Smart DJ, Davis PG . Prophylactic methylxanthines for endotracheal extubation in preterm infants. Cochrane Database Syst Rev 2010: CD000139.
Bhaskara S, Dean ED, Lam V, Ganguly R . Induction of two cytochrome P450 genes, Cyp6a2 and Cyp6a8, of Drosophila melanogaster by caffeine in adult flies and in cell culture. Gene 2006; 377: 56–64.
Misra JR, Horner MA, Lam G, Thummel CS . Transcriptional regulation of xenobiotic detoxification in Drosophila. Genes Dev 2011; 25: 1796–1806.
Cauli O, Morelli M . Caffeine and the dopaminergic system. Behav Pharmacol 2005; 16: 63–77.
Halder G, Johnson RL . Hippo signaling: growth control and beyond. Development 2011; 138: 9–22.
Yu FX, Guan KL . The Hippo pathway: regulators and regulations. Genes Dev 2013; 27: 355–371.
Goulev Y, Fauny JD, Gonzalez-Marti B, Flagiello D, Silber J, Zider A . SCALLOPED interacts with YORKIE, the nuclear effector of the hippo tumor-suppressor pathway in Drosophila. Curr Biol 2008; 18: 435–441.
Wu S, Liu Y, Zheng Y, Dong J, Pan D . The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev cell 2008; 14: 388–398.
Zhang L, Ren F, Zhang Q, Chen Y, Wang B, Jiang J . The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev cell 2008; 14: 377–387.
Peng HW, Slattery M, Mann RS . Transcription factor choice in the Hippo signaling pathway: homothorax and yorkie regulation of the microRNA bantam in the progenitor domain of the Drosophila eye imaginal disc. Genes Dev 2009; 23: 2307–2319.
Oh H, Irvine KD . Cooperative regulation of growth by Yorkie and Mad through bantam. Dev cell 2011; 20: 109–122.
Oh H, Irvine KD . Yorkie: the final destination of Hippo signaling. Trends Cell Biol 2010; 20: 410–417.
Genevet A, Wehr MC, Brain R, Thompson BJ, Tapon N . Kibra is a regulator of the Salvador/Warts/Hippo signaling network. Dev cell 2010; 18: 300–308.
Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C et al. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol 2006; 8: 27–36.
Baumgartner R, Poernbacher I, Buser N, Hafen E, Stocker H . The WW domain protein Kibra acts upstream of Hippo in Drosophila. Dev cell 2010; 18: 309–316.
Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D . Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev cell 2010; 18: 288–299.
Bennett FC, Harvey KF . Fat cadherin modulates organ size in Drosophila via the Salvador/Warts/Hippo signaling pathway. Curr Biol 2006; 16: 2101–2110.
Robinson BS, Huang J, Hong Y, Moberg KH . Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein Expanded. Curr Biol 2010; 20: 582–590.
Chen CL, Gajewski KM, Hamaratoglu F, Bossuyt W, Sansores-Garcia L, Tao C et al. The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc Natl Acad Sci USA 2010; 107: 15810–15815.
Ling C, Zheng Y, Yin F, Yu J, Huang J, Hong Y et al. The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc Natl Acad Sci USA 2010; 107: 10532–10537.
Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D . Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013; 154: 1342–1355.
Cho E, Feng Y, Rauskolb C, Maitra S, Fehon R, Irvine KD . Delineation of a Fat tumor suppressor pathway. Nat Genet 2006; 38: 1142–1150.
Silva E, Tsatskis Y, Gardano L, Tapon N, McNeill H . The tumor-suppressor gene fat controls tissue growth upstream of expanded in the hippo signaling pathway. Curr Biol 2006; 16: 2081–2089.
Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, Sasaki H et al. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J 2011; 30: 2325–2335.
Fernandez BG, Gaspar P, Bras-Pereira C, Jezowska B, Rebelo SR, Janody F . Actin-Capping Protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development 2011; 138: 2337–2346.
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M et al. Role of YAP/TAZ in mechanotransduction. Nature 2011; 474: 179–183.
Wada K, Itoga K, Okano T, Yonemura S, Sasaki H . Hippo pathway regulation by cell morphology and stress fibers. Development 2011; 138: 3907–3914.
Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 2000; 101: 91–101.
Fan Y, Lee TV, Xu D, Chen Z, Lamblin AF, Steller H et al. Dual roles of Drosophila p53 in cell death and cell differentiation. Cell Death Differ 2010; 17: 912–921.
Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM . Drosophila p53 binds a damage response element at the reaper locus. Cell 2000; 101: 103–113.
Basak S, Jacobs SB, Krieg AJ, Pathak N, Zeng Q, Kaldis P et al. The metastasis-associated gene Prl-3 is a p53 target involved in cell-cycle regulation. Mol Cell 2008; 30: 303–314.
Pagarigan KT, Bunn BW, Goodchild J, Rahe TK, Weis JF, Saucedo LJ . Drosophila PRL-1 is a growth inhibitor that counteracts the function of the Src oncogene. PLoS One 2013; 8: e61084.
Liang F, Liang J, Wang WQ, Sun JP, Udho E, Zhang ZY . PRL3 promotes cell invasion and proliferation by down-regulation of Csk leading to Src activation. J Biol Chem 2007; 282: 5413–5419.
King-Jones K, Horner MA, Lam G, Thummel CS . The DHR96 nuclear receptor regulates xenobiotic responses in Drosophila. Cell Metab 2006; 4: 37–48.
Yu J, Pacifico S, Liu G, Finley RL Jr . DroID: the Drosophila Interactions Database, a comprehensive resource for annotated gene and protein interactions. BMC Genomics 2008; 9: 461.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013; 41: D808–D815.
Ribeiro PS, Josue F, Wepf A, Wehr MC, Rinner O, Kelly G et al. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol Cell 2010; 39: 521–534.
Stewart RA, Li DM, Huang H, Xu T . A genetic screen for modifiers of the lats tumor suppressor gene identifies C-terminal Src kinase as a regulator of cell proliferation in Drosophila. Oncogene 2003; 22: 6436–6444.
Colombani J, Polesello C, Josue F, Tapon N . Dmp53 activates the Hippo pathway to promote cell death in response to DNA damage. Curr Biol 2006; 16: 1453–1458.
Ilanges A, Jahanshahi M, Balobin DM, Pfleger CM . Alcohol interacts with genetic alteration of the hippo tumor suppressor pathway to modulate tissue growth in Drosophila. PLoS One 2013; 8: e78880.
Feng Y, Irvine KD . Processing and phosphorylation of the Fat receptor. Proc Natl Acad Sci USA 2009; 106: 11989–11994.
Watabe M, Kakeya H, Osada H . Requirement of protein kinase (Krs/MST) activation for MT-21-induced apoptosis. Oncogene 1999; 18: 5211–5220.
Watabe M, Kakeya H, Onose R, Osada H . Activation of MST/Krs and c-Jun N-terminal kinases by different signaling pathways during cytotrienin A-induced apoptosis. J Biol Chem 2000; 275: 8766–8771.
Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007; 130: 1120–1133.
Oh H, Irvine KD . In vivo regulation of Yorkie phosphorylation and localization. Development 2008; 135: 1081–1088.
Schoenherr JA, Drennan JM, Martinez JS, Chikka MR, Hall MC, Chang HC et al. Drosophila activated Cdc42 kinase has an anti-apoptotic function. PLoS Genet 2012; 8: e1002725.
Li X, Zhuo R, Tiong S, Di Cara F, King-Jones K, Hughes SC et al. The Smc5/Smc6/MAGE complex confers resistance to caffeine and genotoxic stress in Drosophila melanogaster. PLoS One 2013; 8: e59866.
Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D et al. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell 2007; 27: 962–975.
Strano S, Monti O, Pediconi N, Baccarini A, Fontemaggi G, Lapi E et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol Cell 2005; 18: 447–459.
Lapi E, Di Agostino S, Donzelli S, Gal H, Domany E, Rechavi G et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol Cell 2008; 32: 803–814.
Basu S, Totty NF, Irwin MS, Sudol M, Downward J . Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell 2003; 11: 11–23.
Danovi SA, Rossi M, Gudmundsdottir K, Yuan M, Melino G, Basu S . Yes-associated protein (YAP) is a critical mediator of c-Jun-dependent apoptosis. Cell Death Differ 2008; 15: 217–219.
Oka T, Mazack V, Sudol M . Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J Biol Chem 2008; 283: 27534–27546.
Bertini E, Oka T, Sudol M, Strano S, Blandino G . YAP: at the crossroad between transformation and tumor suppression. Cell Cycle 2009; 8: 49–57.
Goyal L, McCall K, Agapite J, Hartwieg E, Steller H . Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J 2000; 19: 589–597.
Bond D, Foley E . A quantitative RNAi screen for JNK modifiers identifies Pvr as a novel regulator of Drosophila immune signaling. PLoS Pathog 2009; 5: e1000655.
Saldanha AJ . Java Treeview—extensible visualization of microarray data. Bioinformatics 2004; 20: 3246–3248.
Genevet A, Polesello C, Blight K, Robertson F, Collinson LM, Pichaud F et al. The Hippo pathway regulates apical-domain size independently of its growth-control function. J Cell Sci 2009; 122: 2360–2370.
Oldham S, Stocker H, Laffargue M, Wittwer F, Wymann M, Hafen E . The Drosophila insulin/IGF receptor controls growth and size by modulating PtdInsP(3) levels. Development 2002; 129: 4103–4109.
Huang J, Wu S, Barrera J, Matthews K, Pan D . The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005; 122: 421–434.
Tomlinson V, Gudmundsdottir K, Luong P, Leung KY, Knebel A, Basu S . JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis 2010; 1: e29.
Levy D, Adamovich Y, Reuven N, Shaul Y . Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell 2008; 29: 350–361.
Johnson R, Halder G . The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 2014; 13: 63–79.
Harvey KF, Zhang X, Thomas DM . The Hippo pathway and human cancer. Nature Rev Cancer 2013; 13: 246–257.
Acknowledgements
We would like to thank the Bloomington Drosophila Stock Center at Indiana University for supplying fly stocks and the RNAi Screening facility at Li Ka-Shing Institute of Virology, University of Alberta. We also would like to thank Edan Foley for his help and advice he provided for the RNAi screening procedures. Finally, we wish to thank the Canadian Institutes of Health Research (CIHR) and the Natural Sciences and Engineering Research Council of Canada (NSERC) for supporting this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article

Cite this article
Di Cara, F., Maile, T., Parsons, B. et al. The Hippo pathway promotes cell survival in response to chemical stress. Cell Death Differ 22, 1526–1539 (2015). https://doi.org/10.1038/cdd.2015.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.10
This article is cited by
-
How do phosphodiesterase-5 inhibitors affect cancer? A focus on glioblastoma multiforme
Pharmacological Reports (2022)
-
p53 shades of Hippo
Cell Death & Differentiation (2018)
