
Abstract
The death inducer obliterator (Dido) locus encodes three protein isoforms, of which Dido3 is the largest and most broadly expressed. Dido3 is a nuclear protein that forms part of the spindle assembly checkpoint (SAC) and is necessary for correct chromosome segregation in somatic and germ cells. Here we report that specific ablation of Dido3 function in mice causes lethal developmental defects at the onset of gastrulation. Although these defects are associated with centrosome amplification, spindle malformation and a DNA damage response, we provide evidence that embryonic lethality of the Dido3 mutation cannot be explained by its impact on chromosome segregation alone. We show that loss of Dido3 expression compromises differentiation of embryonic stem cells in vitro and of epiblast cells in vivo, resulting in early embryonic death at around day 8.5 of gestation. Close analysis of Dido3 mutant embryoid bodies indicates that ablation of Dido3, rather than producing a generalized differentiation blockade, delays the onset of lineage commitment at the primitive endoderm specification stage. The dual role of Dido3 in chromosome segregation and stem cell differentiation supports the implication of SAC components in stem cell fate decisions.
Similar content being viewed by others


Main
The death inducer obliterator (Dido) gene has been described only in higher vertebrates and encodes three splice variants. Dido1, the smallest of these, was identified by differential display PCR in murine bone marrow cells undergoing apoptosis and its ectopic overexpression was shown to induce caspase-dependent cell death.1 Cloning of the Dido gene yielded two further Dido isoforms, Dido2 and Dido3; misexpression of these splice variants was linked to myelodysplastic syndrome/myeloproliferative disease.2 The finding that N-terminal truncation of Dido3 (Dido3ΔNT) provokes increased incidence of hematological myeloid neoplasms in the adult mouse further supports a role for this gene in tumor suppression.2
Subsequent studies have focused on Dido3, the largest and most broadly expressed isoform. Dido3 is a nuclear protein that recognizes trimethylated histone H3 lysine 4 through its N-terminal plant homeodomain domain,3 and interacts with centrosomes and the synaptonemal complex in somatic4 and germ cells,3 respectively. Cells expressing an N-terminally truncated and partially inactive Dido3 form that lacks the sequence motifs needed for association with histones (Dido3ΔNT) show centrosome amplification, spindle malformation and chromosome segregation defects.4 Dido3ΔNT cells bypass the spindle assembly checkpoint (SAC) through unscheduled degradation of BubR1, which renders them permissive to aneuploidy, chromosomal instability and DNA damage.4, 5 A limitation for evaluation of specific Dido3 function in the Dido3ΔNT mouse mutant is that elimination of the sequence encoding the Dido gene N-terminus in the germline also affects Dido1 and Dido2 isoforms. Moreover, elimination of this N-terminal sequence inactivates Dido3 only partially, leaving intact a number of functional domains thought to be involved in Dido3 interaction with DNA, chromatin and other proteins.6, 7 To further explore the role of Dido3, we specifically ablated Dido3 expression in the mouse and established Dido3 mutant embryonic stem (ES) cells. We show that loss of Dido3 expression is embryonic lethal and compromises lineage commitment of ES cells in vitro and of epiblast cells at the onset of gastrulation in vivo.
Results
Ablation of Dido3 is embryonic lethal
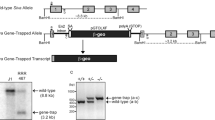
We generated a Dido3-specific mouse mutant using red fluorescence protein (RFP) to replace the only Dido3-specific exon in the Dido locus, which eliminates almost 50% of the Dido3 C-terminal sequence, leaving intact the functional domains in Dido2 (Dido3ΔCT-RFP, Figure 1a; Supplementary Figure 1). The mutant Dido3 allele was transmitted to the germline and yielded macroscopically normal mice heterozygous for full-length Dido3; we were nonetheless unable to generate viable homozygous Dido3ΔCT-RFP mice by intercrossing heterozygotes. Analysis of blastocyst explants and embryos from Dido3 heterozygote intercrosses showed Dido3 mutant embryos at the expected Mendelian frequency up to day 8.5 post-coitum (E8.5), none of which survived beyond this time (Figure 1b). Absence of Dido3 expression in the developing embryo (Supplementary Figure 2) was associated with gross morphological abnormalities that became overt by E7.5 and particularly affected the embryonic ectoderm (Figure 1c). Histological analysis showed that the trophoblast and extraembryonic regions were less affected than the epiblast by Dido3 ablation (Figure 1d). In these embryos, we assessed the ablation of Dido3 and expression of the Dido3ΔCT-RFP mutant by western blot (Figure 1e); we also used reverse transcriptase (RT)-PCR to verify normal expression of Dido1 and Dido2, ablation of Dido3 and expression of the Dido3ΔCT-RFP mutant (Figure 1f). Although we cannot rule out that the Dido3ΔCT-RFP allele might cause a subtle neomorphic phenotype, these data support the interpretation that elimination of the C-terminal portion of Dido3 compromises Dido3 function, resulting in early embryonic lethality.

Ablation of Dido3 is embryonic lethal. (a) The three isoforms encoded by the Dido locus (Dido1, Dido2 and Dido3), as well as the two Dido loss-of-function mutants studied so far (DidoΔNT, previously published;2 Dido3ΔCT-RFP, this study). Common and isoform-specific portions of the Dido proteins are indicated (light gray, regions common to all three Dido isoforms; dark gray, regions common to Dido2 and Dido3; black, Dido3-specific regions; hatching, short isoform-specific C-terminal fragments). The location of the in-frame RFP cassette in the Dido3ΔCT-RFP mutant is indicated (RFP). Numbers denote amino acid position, counted from the N-terminus of the three Dido isoforms. Abbreviations below indicate the approximate positions of Dido functional domains (NLS, nuclear localization signal; PHD, plant homeodomain; TFIIS2M, transcription factor S domain II module; SPOC, Spen paralog and ortholog C-terminal domain; PRO, proline-rich domain; ALA, alanine-rich domain; CC, coiled-coil domain; ARG, arginine-rich domain). (b) Ablation of Dido3 is embryonic lethal. Embryos from intercrosses of Dido3ΔCT-RFP heterozygous mice were genotyped by PCR at different gestational stages. Embryonic days (E) are days post-coitum. Wt/Wt, Wt for Dido3; Wt/ΔCT, heterozygous for Dido3ΔCT-RFP; ΔCT/ΔCT, homozygous for Dido3ΔCT-RFP; ND, not determined due to embryo resorption. An asterisk indicates the presence of overt morphological alterations. (c) Comparison of whole Wt/Wt and ΔCT/ΔCT embryos at E7.5. Regions corresponding to the trophoblast (tr), extraembryonic ectoderm (exe) and embryonic ectoderm (ee) are indicated. (d) Histological comparison of whole Wt/Wt and ΔCT/ΔCT embryos. HE-stained sections of Wt/Wt and ΔCT/ΔCT embryos at E7.5, E8.0 and E8.5 are shown at indicated magnifications. Embryonic regions correspond to tr, exe and ee, as well as head (h) and somites (s). (e) Lysates of Wt/Wt and ΔCT/ΔCT embryos at E7.5 were analyzed by western blot with an anti-Dido3 C-terminus-specific antibody. β-actin was used as loading control (left). The anti-common amino-terminal segment antibody to the N-terminal portion of Dido, common to all Dido isoforms,3 was used to assay lysates of Wt/Wt, Wt/ΔCT and ΔCT/ΔCT embryos at E7.5 for expression of Dido3 and Dido3ΔCT-RFP (right). (f) RT-PCR was used to monitor Wt/Wt and ΔCT/ΔCT E7.5 embryos for expression of the splice variants Dido1, Dido2, Dido3 (1, 2 and 3) and Dido3ΔCT-RFP (ΔCT) using β-actin as control (C)
Abrogation of Dido3 causes chromosome segregation defects and provokes a DNA damage response
Previous work showed that N-terminal truncation of Dido3 (Dido3ΔNT, Figure 1a) impairs the SAC machinery, provoking centrosome amplification, spindle malformation and aneuploidy.3, 4 In cells obtained from Dido3ΔCT-RFP compared with wild type (Wt) embryos at E7.5, we observed an increase in cells with less than one or more than two centrosomes per cell, resulting in a moderate but significant increase in cells with an abnormal centrosome number (Figure 2b).

Abrogation of Dido3 causes chromosome segregation defects and provokes a DNA damage response. (a) Centrosome number was determined by staining for γ-tubulin and centrin (red dots) on cytospin slides of disaggregated Wt/Wt and ΔCT/ΔCT embryos at E7.5. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue background). (b) The incidence of centrosome anomalies in these embryos was quantified as the presence of more than two or less than one centrosome per cell as a percentage of total cells analyzed. Data were obtained from four mice per genotype and analyzed for statistically significant differences using the Fisher exact test. (c) Sections from Wt/Wt and ΔCT/ΔCT embryos at E7.5 were assayed for DNA damage (cells positive for phosphorylated γ-H2AX, brown dots) and apoptosis (TUNEL-positive cells, green dots). Arrowheads indicate areas of positive staining in the epiblast. Nuclei were DAPI-counterstained (blue). DAPI and TUNEL staining were performed on the same sections of a paraffin-embedded embryo, whereas γ-H2AX detection was performed in a different embryo. (d) Sections from Wt/Wt and ΔCT/ΔCT embryos at E7.5 were assayed for proliferation by monitoring BrdU incorporation 1 h post-BrdU injection into pregnant mice, followed by fluorescein isothiocyanate-anti-BrdU staining (green). Nuclei were DAPI stained (blue). Selected areas of the epiblast (indicated by numbers 1 and 3 in Wt/Wt and ΔCT/ΔCT embryos, respectively) and trophoblast (2 and 4 in Wt/Wt and ΔCT/ΔCT embryos, respectively) are shown at higher magnification. (e) p21 and p16 expression levels were assayed by RT-PCR in tissue homogenates of Wt/Wt and ΔCT/ΔCT embryos at E7.5. Data show mean±S.E.M. from three mice per genotype, each in triplicate and normalized to 18S mRNA expression. Statistical significance was calculated using Student's t-test. (f) Representative western blot analysis of Wt/Wt and ΔCT/ΔCT embryo lysates at E7.5 for the active growth-promoting form of retinoblastoma protein phosphorylated at serines 807/811 (pRb), using β-actin as loading control
Chromosome segregation defects are a potential source of DNA damage and chromosomal instability.8 In Dido3ΔCT-RFP E7.5 embryos, we detected cells that stained positive for phosphorylated γ-H2AX, a surrogate marker for DNA damage (Figure 2c, top). DNA damage in the Dido3 mutants was paralleled by an apoptotic response, indicated by accumulation of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells (Figure 2c, bottom). Centrosome dysfunction is also linked to a p53/p21-dependent block of cell cycle progression.9 We found that proliferation rates, measured as bromodeoxyuridine (BrdU) incorporation into embryonic tissues, were lower in Dido3ΔCT-RFP embryos than in Wt controls (Figure 2d). Both apoptosis and growth arrest were observed in the epiblast region, and affected extraembryonic tissue only to a minor extent (Figures 2c and d). Impaired proliferation in Dido3-deficient embryos was accompanied by a significant increase in p21 and p16 expression (Figure 2e), as well as by hypophosphorylation of the retinoblastoma protein (Figure 2f). These molecular changes are reminiscent of a canonical senescence response to DNA damage, which requires activation of the p53/p21 and p16/retinoblastoma (Rb) pathways.10 Nonetheless, when we crossed the Dido3ΔCT mutation onto a p53-deficient background, a genetic manipulation intended to render Dido3 mutants resistant to p53-dependent apoptosis and growth arrest, we were unable to rescue the lethal phenotype of Dido3ΔCT-RFP mice (Supplementary Table 1).
Ablation of Dido3 disrupts epiblast cell differentiation
As shown above, accumulation of apoptotic and growth-arrested epiblast cells in Dido3ΔCT-RFP mutants at E7.5 coincides with the onset of developmental defects in the embryonic ectoderm. Epiblast integrity is critical to early embryonic development, as this region contains pluripotent cells from which all three embryo layers are derived during gastrulation.11 Epiblast cell differentiation is triggered by repression of transcription factors necessary for establishing and maintaining stem cell pluripotency, such as Oct4.12 We found that by E7.5, Oct4 was no longer detectable in Wt embryos, concurring with the literature,13 whereas Dido3 mutant embryos showed persistent Oct4 expression in the epiblast (Figure 3a). Brachyury, a specific marker for nascent mesoderm,14 was clearly expressed in Wt embryos but virtually absent from Dido3 mutants, whereas the diploid trophoblast marker Mash-215 was observed in both embryo types (Figure 3b). Dido3ΔCT-RFP embryos also showed altered expression of mRNA encoding the mesodermal marker proteins Brachyury and Twist, the visceral endoderm marker HNF4 and the anterior patterning markers Nodal and Cerberus1 (Figure 3c). These findings indicate that Dido3 is essential for an early stage of embryogenesis related to lineage commitment at the onset of epiblast cell differentiation.

Lack of Dido3 expression disrupts epiblast cell differentiation. (a) Whole embryos from Wt/Wt and ΔCT/ΔCT at E7.5 were examined by immunofluorescence for expression of Oct4, an undifferentiated pluripotent cell marker. Diffuse green staining in the Wt embryo indicates background (lack of Oct4 expression). Arrowheads mark regions with persistent Oct4 expression in the epiblast of a representative ΔCT/ΔCT embryo. Embryonic regions correspond to tr, exe and ee. (b) In situ hybridization was used to assess expression of Brachyury and Mash2, markers specific for nascent mesoderm and diploid trophoblast cells, respectively, in sections from Wt/Wt and ΔCT/ΔCT embryos at E7.5. Arrowheads indicate regions that stain positive for these markers. (c) RT-PCR analysis was used to quantify expression of the mesodermal markers Twist and Brachyury, the visceral endoderm marker HNF4 and the anterior patterning gene products Nodal and Cerberus1 (Cer1). Data are mean values±S.E.M., n=3
Dido3 is dispensable for ES cell self-renewal and pluripotency
The embryonic post-implantation phase (up to E6.5) is characterized by rapid proliferation of stem cells derived from the inner cell mass. By the beginning of gastrulation (E6.5), stem cell proliferation ceases and differentiation into the three germ layers is initiated.16 As this early stage of stem cell differentiation was associated with the onset of lethal developmental defects in Dido3 mutants, we established ES cell lines from blastocyst outgrowths obtained from intercrosses of Dido3ΔCT-RFP heterozygotes and analyzed their capacity to proliferate and differentiate in vitro. As predicted by the presence of cells with chromosome segregation defects in Dido3ΔCT-RFP embryos, Dido3ΔCT-RFP ES cells were aneuploid (Figure 4a), with centrosome amplification and multipolar spindles in ∼10% of cells (Figure 4b). Despite these chromosome segregation defects, the proliferative capacity of undifferentiated Dido3 mutant ES cells was unaffected (Figure 4c, top), indicating that Dido3 is dispensable for ES cell self-renewal. To explore the impact of Dido3 abrogation on ES cell differentiation, we used the hanging drop method to aggregate ES cells into embryonic bodies (EBs) and induced their further differentiation by propagation in gelatinized cell culture dishes in the absence of leukemia inhibitory factor (LIF). In these conditions, differentiation of Dido3ΔCT-RFP ES cells was impaired and they underwent growth arrest and apoptosis (Figure 4c, bottom), which raises the question whether loss of Dido3 expression abrogates ES cell pluripotency. Teratoma assays are widely used to demonstrate the differentiation potential of pluripotent ES cells.17 We found that subcutaneous injection of Dido3ΔCT-RFP ES cells into immunodeficient mice yielded teratomas (Figure 4d), although at a significantly lower frequency than Wt ES cells (Figure 4e). Histological analysis of Dido3 mutant teratomas nonetheless confirmed that Dido3ΔCT-RFP ES cells retained the capacity of pluripotent ES cells to form all three germ layers after ectopic transplant (Figure 4f). These results suggest that abrogation of Dido3 expression, although it impairs ES cell differentiation in vitro, does not produce a generalized blockade of lineage commitment in vivo.

Dido3 is dispensable for ES cell self-renewal and pluripotency. (a) Colcemid-arrested metaphase spreads from Wt/Wt and ΔCT/ΔCT ES cells were karyotyped. Cells with more or less than 40 chromosomes (2N) were scored as aneuploid. The number of aneuploid metaphases relative to the total number of metaphases analyzed is shown above each bar. The Fisher exact test was used for statistical comparison. (b) Two representative examples for the presence of multipolar spindles (γ-tubulin; red dots) in interphase and mitotic nuclei of ΔCT/ΔCT ES cells. (c) Cell cycle profiles of propidium iodide (Pi)-stained Wt/Wt and ΔCT/ΔCT ES cells maintained as undifferentiated ES cells (top) or differentiated in vitro by aggregation into hanging drops and subsequent adhesion of the EB obtained to gelatinized cell culture dishes (bottom). Apoptotic cells (Ap) and cell ploidy (1N, 2N and 4N) are indicated; the inset shows representative photomicrographs for Wt/Wt and ΔCT/ΔCT EB. The proliferative capacity of ES cells and EB was assayed by BrdU incorporation and quantified on scatter-plots of BrdU versus Pi signal. The percentage of proliferating cells is indicated. (d) Pluripotency of Wt/Wt and ΔCT/ΔCT ES cells was assessed by ectopic transplant into immunodeficient recombination activating gene 2 knockout mice (106 cells/injection). Representative examples are shown of teratoma formation after injection of Wt/Wt and ΔCT/ΔCT ES cells. (e) Bar graph quantifies the efficiency of teratoma formation from Wt/Wt and ΔCT/ΔCT ES cells. The number of injections that yielded overt teratomas within 6 weeks relative to total number of injections is given above each bar. The Fisher exact test was used for statistical comparisons. (f) Mice were killed at 6 weeks post-injection to isolate teratomas. Histology of HE-stained ΔCT/ΔCT teratoma sections confirms that ΔCT/ΔCT ES cells retain the capacity to differentiate into all three germ layers (endoderm, mesoderm, ectoderm)
Abrogation of Dido3 compromises ES cell lineage commitment
Aggregation of ES cells into EB and their further differentiation into all three germ layers is considered to be an in vitro model that recapitulates essential aspects of early embryonic development.18 Following aggregation, Wt ES cells formed EB; when they were maintained in suspension culture for several days in the absence of LIF, they continued to differentiate, as evidenced by characteristic changes in cell morphology and loss of alkaline phosphatase (AP) staining (Figure 5a, top). Dido3ΔCT-RFP ES cells retained the capacity to form EB, but did not differentiate further in these conditions (Figure 5a, bottom), determined by persistent AP staining and absence of morphological changes as well as by expression of stem cell genetic markers.

Dido3 is necessary for ES cell differentiation in vitro. Wt/Wt and ΔCT/ΔCT ES cells were cultured with LIF to impede differentiation (ES), or were aggregated into EB and maintained without LIF for the indicated time in untreated bacterial plates to induce differentiation. (a) AP staining was used as an indicator of undifferentiated and pluripotent stem cells. Photomicrographs show representative results for ES cells and EB at × 20 and × 10 magnification, respectively. (b) Western blot analysis was used to assay expression of proteins considered pluripotency markers (Oct4, Nanog, Sox2) in ES cells and EB; β-actin was used as a loading control
There is evidence that the activity of a set of transcriptional regulators, including Oct4, Nanog and Sox2, is sufficient to confer pluripotency and to regulate stem cell self-renewal and differentiation.19 ES cell differentiation is dependent on the progressive downregulation of these transcriptional regulators.12 Oct4, Nanog and Sox2 protein expression in Wt ES cells began to decline 4 to 6 days after EB formation and LIF withdrawal, and was barely detectable by day 14 (Figure 5b, left). In Dido3 mutant EB, Oct4 repression was abrogated and downregulation of Sox2 and Nanog was delayed (Figure 5b, right), confirming that Dido3 is necessary for LIF-dependent ES cell differentiation in vitro.
Although LIF is a key factor in maintaining ES cells in an undifferentiated state, LIF removal is not essential for initial specification of the primitive endoderm induced by ES cell aggregation into EB.20 Aggregation-induced ES cell differentiation in vitro begins with the formation of a primitive endoderm layer on the surface of the ES cell aggregate, which is GATA-4-dependent21 and a prerequisite for basement membrane formation and subsequent separation of the primitive endoderm from the inner EB cell mass.22 After aggregation and subsequent culture without LIF, Wt EB began to form a primitive, GATA-4-expressing endoderm layer; primitive endoderm formation in Wt EB was completed by day 2–3, followed by the onset of ES cell differentiation (Figure 6a). In these conditions, primitive endoderm formation was delayed in Dido3 mutant EB, as evidenced by their morphology and the delayed onset of GATA-4 expression (Figure 6a). We noted a comparable delay in primitive endoderm formation in Dido3ΔCT-RFP EB after aggregation in the presence of LIF; after 5 days, Dido3 mutant ES cells completed formation of a GATA-4-positive primitive endoderm layer (Figure 6b). These data indicate that loss of Dido3 expression delays primitive endoderm specification independent of LIF signaling.

Ablation of Dido3 delays aggregation-induced primitive endoderm formation. Photomicrographs (gray-tone images) show representative EB obtained by aggregation of Wt/Wt and ΔCT/ΔCT ES cells and subsequent propagation in the absence (w/o) (a) or presence (with) (b) of LIF for the time indicated. Primitive endoderm formation was monitored by immunofluorescence using antibody to the endoderm marker GATA-4 (bright red signal)
We performed various experiments to study whether, once primitive endoderm formation was complete, Dido3ΔCT-RFP EB were able to exit from the pluripotent state and to differentiate. In a first assay, we aggregated Dido3ΔCT-RFP ES cells into EB, triggered primitive endoderm formation for 5 days with LIF (see Figure 6a) and subsequently induced differentiation by LIF withdrawal; Dido3ΔCT-RFP EB appeared able to differentiate (Figure 7a). We also co-cultured Wt and mutant ES cells to establish in vitro chimeras. In these conditions, the kinetics of primitive endoderm formation resembled that of the Wt cells and differentiation was similar to that in Wt EB, as detected by the expression of specific genes (not shown). Finally, as defects in primitive endoderm formation in GATA-4-deficient ES cells21 can be rescued by activation of the retinoic acid signaling pathway,23 we tested this system and found that retinoic acid restored Dido3ΔCT-RFP EB ability to differentiate, indicated by morphological changes similar to those of Wt cells (not shown).

Impaired differentiation of Dido3 mutant ES cells can be rescued in vitro. (a) Photomicrographs of representative EB obtained by aggregation of Wt/Wt and ΔCT/ΔCT ES cells and subsequent propagation with LIF for 5 days before inducing differentiation by LIF withdrawal for a further 9 days. (b) Quantitative RT-PCR was used to determine the expression of selected markers for undifferentiated ES cells, endoderm, mesoderm and ectoderm in differentiating EB. Differentiation was induced by aggregation of ES into EB and maintenance of EB for a further 10 days in suspension on untreated bacterial plates in medium without LIF (red bars) and supplemented with 0.5 μM retinoic acid (green bars). Alternatively, differentiation was induced by aggregation of ES cells into EB and subsequent maintenance as in a (blue bars). Data show mean values for two experiments±S.E.M. and represent expression of the markers analyzed in ΔCT/ΔCT EB relative to Wt/Wt EB
To evaluate the impact of experimental conditions that delayed endoderm specification in Dido3 mutant EB, we screened EB for markers representative of undifferentiated ES cells and of all three germ layers. Compared with Wt EB, Dido3 mutant EB cultured for 10 days without LIF showed an overall persistence of genes associated to undifferentiated ES cells (Figure 7b, red bars); this included a three to fivefold increase in pluripotency determinants such as Oct4, Nanog and Foxd3, as well as in genes that prevent cultured ES cell differentiation such as Nodal, Cripto7 and Gdf3.24, 25 Expression of endoderm, mesoderm and ectoderm markers, all indicative of differentiation, was decreased (Figure 7b, red bars). The extraembryonic endoderm genes GATA-4 and GATA-6, necessary for endoderm differentiation,21, 23 were expressed at 10- to 20-fold lower levels in Dido3 mutants than in Wt EB (Figure 7b, red bars). There was an eightfold decrease in expression of the endoderm marker laminin B1 (Lamb1), a protein associated with deposition of subendodermal basement membrane by primitive endoderm cells and necessary for subsequent germ layer formation26 (Figure 7b, red bars). These data confirm that Dido3 ablation impairs stem cell differentiation already at the onset of primitive endoderm formation. Conditions that allow completion of endoderm formation in Dido3ΔCT-RFP EB before induction of differentiation (5 days preliminary culture with LIF) partially restored the Wt expression pattern; this included downregulation of pluripotency-associated genes (Oct4, Sox2) and induction of genes required for differentiation of endoderm (GATA-4/6, Lamb1), mesoderm (Flt1, Cdh5/VE-cadherin) and ectoderm (Syp) (Figure 7b, blue bars). When retinoic acid was used to bypass delayed primitive endoderm formation in Dido3ΔCT-RFP EB, suppression of pluripotency genes and restored expression of genes representing all three germ layers was even more pronounced (Figure 7b, green bars). These findings show that impaired differentiation of Dido3ΔCT-RFP ES cells can be overcome in vitro in conditions that allow prior completion of primitive endoderm specification.
The role of Dido3 in ES cell differentiation can be separated from its function in chromosome segregation
Partial inactivation of Dido gene products, by truncation of the N-terminal domain common to the three Dido isoforms (Dido3ΔNT), impairs the SAC machinery and provokes centrosome amplification, spindle malformation, aneuploidy and DNA damage.4, 5 The extensive Dido3 truncation in the Dido3ΔCT-RFP mutant might exacerbate the effects observed after partial Dido inactivation in the Dido3ΔNT mutant, which could account for impaired stem cell differentiation, developmental defects and early embryonic lethality in Dido3ΔCT-RFP mice. Chromosome imbalance in Dido3ΔCT-RFP mutants is comparable to that in Dido3ΔNT mice, although there is no appreciable impact on development in the latter.4 Mitotic defects thus do not appear to account fully for the lethal phenotype of the Dido3ΔCT-RFP mutation. To test this hypothesis, we generated double mutants by crossing heterozygous Dido3ΔCT-RFP with heterozygous Dido3ΔNT mice; although Dido3ΔCT-RFP/Dido3ΔNT mice were not born at the expected Mendelian frequency, we obtained viable Dido3 double mutants (Figure 8a). Karyotype analysis of Dido3ΔCT-RFP/Dido3ΔNT and Wt embryo primary fibroblasts showed an ∼twofold increase in aneuploid cells (58.7±6.5% versus 28.7±3.7%, n=2), accompanied by an increase in structural chromosomal aberrations and premature sister chromatid exchange (not shown). In in vitro experiments, overexpression of Dido3ΔNT in Dido3ΔCT-RFP ES cells did not rescue centrosome amplification (Figure 8b), but restored the capacity of these cells to express markers representative of all three germ layers (Figure 8c). At difference, overexpression of full-length Dido3 in Dido3ΔCT-RFP ES cells reconstituted both normal centrosome numbers and the ability of cells to differentiate in the absence of LIF. The finding that Dido3ΔNT rescues specific Dido3ΔCT-RFP defects indicates a function of full-length Dido3 in stem cell differentiation that can be segregated from its known role in the SAC.

The role of Dido3 in ES cell differentiation can be distinguished from its function in chromosome segregation. (a) Dido3ΔCT-RFP embryonic lethality was rescued by intercrossing heterozygous Wt/ΔCT with heterozygous Wt/ΔNT mice to obtain a double mutant (ΔCT/ΔNT). Total number of viable pups is shown for each genotype; the contribution of each genotype relative to the total number of mice born is indicated in parentheses. (b) Full-length Dido3 (Dido3) and the N-terminal Dido3 truncation mutant (Dido3ΔNT) were overexpressed in ΔCT/ΔCT ES cells. Centrosome number was determined by staining for γ-tubulin. Cells were scored as positive for centrosome anomalies when more than two or less than one centrosome per cell were detected. The number of cells with centrosome amplification as a percentage of the total number of cells analyzed is shown above each bar. The Fisher exact test was used for statistical comparison with Wt/Wt ES cells. (c) Full-length Dido3 and Dido3ΔNT were overexpressed in ΔCT/ΔCT ES cells. Differentiation was induced by aggregation into EB and maintenance of EB for a further 10 days without LIF. Quantitative RT-PCR was used to determine the expression of selected markers for undifferentiated ES cells, endoderm, mesoderm and ectoderm in ΔCT/ΔCT EB overexpressing full-length Dido3 (red bars) and Dido3ΔNT (blue bars). Data show mean values of two experiments±S.E.M. and represent expression of the markers analyzed in ΔCT/ΔCT EB transfected with Dido3 and Dido3ΔNT, relative to Wt/Wt EB
Discussion
Previous characterization of a mouse model bearing an N-terminal Dido3 truncation mutant (Dido3ΔNT)2 showed an essential role for Dido3 in SAC regulation and surveillance.4 As the hypomorphic Dido3ΔNT mutant conserves functional domains related to cell signaling and transcriptional regulation,6, 7 we speculate that Dido3 has biologically relevant functions beyond its established role in chromosome segregation. By analyzing mice with a Dido3-specific mutation that eliminates almost 50% of the Dido3 C-terminal sequence, we show that Dido3 is essential for early embryonic development.
In accordance with findings in somatic4 and germ3 cells, we observed that absence of full-length Dido3 is associated with multipolar spindles, centrosomal amplification and aneuploidy. We show that these chromosome anomalies provoke DNA damage, apoptosis and growth arrest during ES cell differentiation in vitro and during early embryonic development in vivo. These findings fit well with reports on the phenotype of mice deficient for other regulators of spindle assembly and mitosis, such as BubR1, Bub3, Rae1 and Mad2.27 Although SAC disruption is generally associated with embryonic lethality,27 we provide evidence that abrogation of Dido3 expression disrupts early embryonic development through mechanisms different from those implicated in chromosome segregation. Genetic ablation of p53 failed to render Dido3ΔCT mutants resistant to p53-dependent apoptosis and growth arrest. Moreover, genetic rescue experiments showed that Dido3ΔNT was unable to reduce chromosome segregation defects in Dido3ΔCT-RFP cells, but rescued embryonic lethality of the Dido3ΔCT mutation. We further show that differentiation is impaired in Dido3ΔCT-RFP ES cells and that Dido3ΔNT overexpression rescues this defect. As Dido3 is ubiquitously expressed in the developing embryo, we cannot rule out that abrogation of Dido3 affects regions other than the epiblast. Nonetheless, the observation that onset of embryonic lethality in Dido3ΔCT-RFP mice coincides with failure of epiblast cell differentiation supports our conclusion that Dido3 expression is crucial in stem cell differentiation.
Genome-wide screens have identified a number of potential regulators of stem cell function, including genes with established roles in transcriptional regulation, cell growth and differentiation, but also others whose function is largely unknown or remains to be validated, such as the Dido gene.28, 29, 30, 31, 32, 33 Here we use the model of ES cell aggregation into EB and subsequent LIF withdrawal to show that Dido3ΔCT-RFP ES cells do not differentiate correctly. Loss of Dido3 expression nonetheless does not produce a generalized differentiation blockade, corroborated by the observation that Dido3ΔCT-RFP ES cells differentiate in response to retinoic acid and form all three germ layers in the teratoma assay. The Dido3 mutant ES cell phenotype is reminiscent of GATA-4-deficient ES cells, a model for disrupted endoderm lineage commitment.21, 34 Like Dido3 mutants, GATA-4-deficient ES cells fail to differentiate after aggregation and LIF withdrawal, as shown by sustained expression of Oct4 and other undifferentiated stem cell markers; their ability to differentiate can be restored by retinoic acid.21, 23, 34 GATA-4 expression is needed in certain signaling pathways to detect cell aggregation and cell positioning, and thus induce differentiation; this requirement can be bypassed by retinoic acid.23 Dido3ΔCT-RFP ES cells recover their capacity to differentiate in response to LIF withdrawal when the previous aggregation period is prolonged and the mutants complete primitive endoderm formation. This suggests that, among its other possible effects, loss of Dido3 expression impairs ES cell differentiation by delaying primitive endoderm specification. We propose that this delayed onset of stem cell differentiation, although it can be bypassed in certain experimental conditions in vitro (retinoic acid treatment, prolonged aggregation) and in vivo (teratoma assay), explains impaired epiblast differentiation and contributes to embryonic lethality in the Dido3ΔCT-RFP mouse model.
How lack of Dido3 expression delays primitive endoderm formation remains to be determined. The initial defect could be caused by the delayed downregulation of stemness-associated transcription factors (particularly in the case of Oct4), by delayed upregulation of genes needed to form primitive endoderm, or by a combination of both. We distinguished the role of full-length Dido in chromosome segregation from the specific function of the Dido3 C terminus in stem cell differentiation; however, this does not rule out a functional link between the two processes. A recent study shows that loss of expression of BubR1, a key SAC component, compromises the differentiation potential of adult stem cells.35 Correct spindle positioning ensures proper chromosome inheritance to daughter cells during each cell division. Definition of the cleavage plane by correct spindle pole positioning and close surveillance of this process by the SAC36 are the basis of asymmetric stem cell division and differentiation.37, 38 Adhesion molecules are thought to enable the fundamental stem cell property of switching between asymmetrical and symmetrical division by regulating centrosome positioning and spindle angle in response to cell–cell contacts between the stem cell and cells in its niche.39 The role of Dido3 in the SAC,4 which senses and signals centrosome position and spindle tension,36 and its implication in an early cell–cell contact-induced ES cell differentiation step, render Dido3 an appropriate candidate for future study of proteins that couple centrosome orientation to stem cell fate decisions.
Materials and Methods
Mice and ES cells
Dido3 mutant mice were generated by targeting the only Dido3-specific exon (exon 16) of the Dido locus. Details of gene targeting strategy are available online as Supplementary Information (Supplementary Figure 1). Mice were handled in accordance with national and European Union legislation, with approval of the Committee for Ethics in Animal Experimentation of the Centro Nacional de Biotecnología (CNB/CSIC).
To establish ES cell lines, blastocysts were flushed from the uterus of pregnant female mice at 3.5 days post-coitum and immediately plated in IVF dishes on irradiated mouse embryonic fibroblasts (MEF) in ES cell medium. Inner cell mass outgrowths were picked after 5–7 days, fragmented following limited digestion in a drop of 0.25% trypsin/EDTA and replated into individual wells of 12-well plates on a layer of irradiated MEF. For genotyping, MEF were removed from the cultures and ES cells were passaged three times on gelatin-coated plates. ES cells were maintained in DMEM (Gibco, Invitrogen, Carlsbad, CA, USA) with 20% FCS, non-essential amino acids, 0.05 mM 2-mercaptoethanol, 100 U/ml penicillin, 0.1 mg/ml streptomycin and 1000 U/ml murine LIF (Chemicon International, Temecula, CA, USA) on a layer of irradiated or mitomycin C-treated MEF. For embryoid body formation, 103 ES cells/0.03 ml LIF-free culture medium were suspended in hanging drops and maintained for 2–3 days, and/or transferred to uncoated Petri dishes and cultured in suspension in the same medium.
RNA and protein analysis
Total RNA was prepared from ES cell or whole embryo lysates using Trizol (Invitrogen). RNA was analyzed by RT-PCR using Superscript III (Invitrogen) according to standard procedures. Quantitative real-time RT-PCR was performed using SYBR green and specific primers (sequences available from the authors on request).
For western blot analysis of protein expression, embryos, ES cells or EB were boiled in 2 × sample buffer containing SDS and mercaptoethanol (Merck, Darmstadt, Germany). Protein extracts were cleared by centrifugation, separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose. Blot membranes were blocked in Tris-buffered saline solution with Tween20-5% non-fat dry milk and probed with anti-Dido3 (generated in our lab), -pRb (1 : 1000; 9308, Cell Signaling, Beverly, MA, USA), -Oct4 (1 : 200; sc-5279, Santa Cruz Biotechnology, Santa Cruz, CA, USA), -Nanog (1 : 1000; AB9220, Chemicon International), -Sox2 (1 : 500; AB5603, Chemicon International) or -actin antibody (1 : 5000; AC-74, Sigma, St Louis, MO, USA).
For immunofluorescence, ES cells, cell explants from trypsin/EDTA-digested (30–45 min, 37°C) embryos, whole embryos or EB were fixed with 4% paraformaldehyde in PBS, permeabilized with 1% Triton X-100 and stained with antibodies to centrin1 (1 : 200; ab 11257, Abcam, Cambridge, UK), γ-tubulin (1 : 1000; T 6557, Sigma), γ-H2AX (1 : 250; IHC-00059, Bethyl Laboratories, Montgomery, TX, USA), Oct4 (1 : 50; Santa Cruz) or GATA-4 (1 : 40; AF2606, R&D Systems, Minneapolis, MN, USA). ES cells and EB were stained for AP with the StemTAG kit (Cell Biolabs, San Diego, CA, USA).
For whole-embryo analysis, uteruses were isolated at the indicated days post-coitum, fixed in 4% paraformaldehyde (4 h), dehydrated, paraffin-embedded and sectioned (0.05 mm for hematoxylin/eosin (HE) staining or immunohistochemistry; 0.01 mm for in situ hybridization). Sections were incubated on slides (37°C, overnight), deparaffinized, rehydrated and used for HE staining, immunohistochemistry or in situ hybridization. For anti-BrdU-fluorescein isothiocyanate labeling of S-phase cells, pregnant females received an injection of BrdU (0.1 mg/g body weight) 1 h before being killed, uteruses were removed and treated as above. We detected apoptotic cells by the TUNEL assay using the In Situ Cell Death Detection kit (Roche, Mannheim, Germany).
Dido3 expression constructs and transfection
For reconstitution experiments, we cloned cDNA of full-length Dido3 and N-terminal truncated Dido3 into the pCAGGS mammalian expression vector in front of an internal ribosome entry site and puromycin-resistance cassette. ES cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol; 24 h after transfection, puromycin (1 μg/ml) was added to culture medium and cells were selected for puromycin resistance for at least 1week. Pooled colonies and/or single colonies were picked, expanded and tested for overexpression of full-length Dido3 or ΔNTDido3.
Abbreviations
- AP:
-
alkaline phosphatase
- BrdU:
-
bromodeoxyuridine
- Dido:
-
death inducer obliterator
- EB:
-
embryonic bodies
- ES:
-
embryonic stem
- HE:
-
hematoxylin/eosin
- Lamb1:
-
laminin B1
- LIF:
-
leukemia inhibitory factor
- MEF:
-
mouse embryonic fibroblasts
- Pi:
-
propidium iodide
- Rb:
-
retinoblastoma
- RFP:
-
red fluorescence protein
- RT-PCR:
-
reverse transcriptase PCR
- SAC:
-
spindle assembly checkpoint
- TUNEL:
-
terminal deoxynucleotidyl transferase dUTP nick end labeling
- Wt:
-
wild type
References
Garcia-Domingo D, Leonardo E, Grandien A, Martinez P, Albar JP, Izpisua-Belmonte JC et al. DIO-1 is a gene involved in onset of apoptosis in vitro, whose misexpression disrupts limb development. Proc Natl Acad Sci USA 1999; 96: 7992–7997.
Futterer A, Campanero MR, Leonardo E, Criado LM, Flores JM, Hernandez JM et al. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J Clin Invest 2005; 115: 2351–2362.
Prieto I, Kouznetsova A, Futterer A, Trachana V, Leonardo E, Alonso Guerrero A et al. Synaptonemal complex assembly and H3K4Me3 demethylation determine DIDO3 localization in meiosis. Chromosoma 2009; 118: 617–632.
Trachana V, van Wely KH, Guerrero AA, Futterer A, Martinez-A C . Dido disruption leads to centrosome amplification and mitotic checkpoint defects compromising chromosome stability. Proc Natl Acad Sci USA 2007; 104: 2691–2696.
Guerrero AA, Gamero MC, Trachana V, Futterer A, Pacios-Bras C, Diaz-Concha NP et al. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc Natl Acad Sci USA 2010; 107: 4159–4164.
Sanchez-Pulido L, Rojas AM, van Wely KH, Martinez-A C, Valencia A . SPOC: a widely distributed domain associated with cancer, apoptosis and transcription. BMC Bioinformatics 2004; 5: 91.
Rojas AM, Sanchez-Pulido L, Futterer A, van Wely KH, Martinez-A C, Valencia A . Death inducer obliterator protein 1 in the context of DNA regulation. Sequence analyses of distant homologues point to a novel functional role. FEBS J 2005; 272: 3505–3511.
Guerrero AA, Martinez-A C, van Wely KH . Merotelic attachments and non-homologous end joining are the basis of chromosomal instability. Cell Div 2010; 5: 13.
Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S . Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat Cell Biol 2007; 9: 160–170.
Collado M, Blasco MA, Serrano M . Cellular senescence in cancer and aging. Cell 2007; 130: 223–233.
Tam PP, Behringer RR . Mouse gastrulation: the formation of a mammalian body plan. Mech Dev 1997; 68: 3–25.
Zernicka-Goetz M, Morris SA, Bruce AW . Making a firm decision: multifaceted regulation of cell fate in the early mouse embryo. Nat Rev Genet 2009; 10: 467–477.
Kaji K, Nichols J, Hendrich B . Mbd3, a component of the NuRD co-repressor complex, is required for development of pluripotent cells. Development 2007; 134: 1123–1132.
Herrmann BG, Kispert A . The T genes in embryogenesis. Trends Genet 1994; 10: 280–286.
Tanaka M, Gertsenstein M, Rossant J, Nagy A . Mash2 acts cell autonomously in mouse spongiotrophoblast development. Dev Biol 1997; 190: 55–65.
Rossant J . Lineage development and polar asymmetries in the peri-implantation mouse blastocyst. Semin Cell Dev Biol 2004; 15: 573–581.
Aleckovic M, Simon C . Is teratoma formation in stem cell research a characterization tool or a window to developmental biology? Reprod Biomed Online 2008; 17: 270–280.
Niwa H . Mouse ES cell culture system as a model of development. Dev Growth Differ 2010; 52: 275–283.
Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005; 122: 947–956.
Hamazaki T, Oka M, Yamanaka S, Terada N . Aggregation of embryonic stem cells induces Nanog repression and primitive endoderm differentiation. J Cell Sci 2004; 117: 5681–5686.
Soudais C, Bielinska M, Heikinheimo M, MacArthur CA, Narita N, Saffitz JE et al. Targeted mutagenesis of the transcription factor GATA-4 gene in mouse embryonic stem cells disrupts visceral endoderm differentiation in vitro. Development 1995; 121: 3877–3888.
Coucouvanis E, Martin GR . Signals for death and survival: a two-step mechanism for cavitation in the vertebrate embryo. Cell 1995; 83: 279–287.
Capo-Chichi CD, Rula ME, Smedberg JL, Vanderveer L, Parmacek MS, Morrisey EE et al. Perception of differentiation cues by GATA factors in primitive endoderm lineage determination of mouse embryonic stem cells. Dev Biol 2005; 286: 574–586.
Hough SR, Laslett AL, Grimmond SB, Kolle G, Pera MF . A continuum of cell states spans pluripotency and lineage commitment in human embryonic stem cells. PLoS One 2009; 4: e7708.
Mesnard D, Guzman-Ayala M, Constam DB . Nodal specifies embryonic visceral endoderm and sustains pluripotent cells in the epiblast before overt axial patterning. Development 2006; 133: 2497–2505.
Murray P, Edgar D . Regulation of laminin and COUP-TF expression in extraembryonic endodermal cells. Mech Dev 2001; 101: 213–215.
Baker DJ, Chen J, van Deursen JM . The mitotic checkpoint in cancer and aging: what have mice taught us? Curr Opin Cell Biol 2005; 17: 583–589.
Kim J, Chu J, Shen X, Wang J, Orkin SH . An extended transcriptional network for pluripotency of embryonic stem cells. Cell 2008; 132: 1049–1061.
Kidder BL, Yang J, Palmer S . Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS One 2008; 3: e3932.
Campbell PA, Perez-Iratxeta C, Andrade-Navarro MA, Rudnicki MA . Oct4 targets regulatory nodes to modulate stem cell function. PLoS ONE 2007; 2: e553.
Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008; 133: 1106–1117.
Brandenberger R, Wei H, Zhang S, Lei S, Murage J, Fisk GJ et al. Transcriptome characterization elucidates signaling networks that control human ES cell growth and differentiation. Nat Biotechnol 2004; 22: 707–716.
Babaie Y, Herwig R, Greber B, Brink TC, Wruck W, Groth D et al. Analysis of Oct4-dependent transcriptional networks regulating self-renewal and pluripotency in human embryonic stem cells. Stem Cells 2007; 25: 500–510.
Bielinska M, Wilson DB . Induction of yolk sac endoderm in GATA-4-deficient embryoid bodies by retinoic acid. Mech Dev 1997; 65: 43–54.
Lee J, Lee CG, Lee KW, Lee CW . Cross-talk between BubR1 expression and the commitment to differentiate in adipose-derived mesenchymal stem cells. Exp Mol Med 2009; 41: 873–879.
Maresca TJ, Salmon ED . Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. J Cell Sci 2010; 123: 825–835.
Gonczy P . Mechanisms of asymmetric cell division: flies and worms pave the way. Nat Rev Mol Cell Biol 2008; 9: 355–366.
Mantel C, Broxmeyer HE . A new connection between the spindle checkpoint, asymmetric cell division and cytokine signaling. Cell Cycle 2007; 6: 144–146.
Marthiens V, Kazanis I, Moss L, Long K, Ffrench-Constant C . Adhesion molecules in the stem cell niche--more than just staying in shape? J Cell Sci 2010; 123: 1613–1622.
Acknowledgements
We thank Drs K van Wely, V Trachana and C Pacios-Bras for critical discussion, L Almonacid of the DIO Genomics Group for low-density array analysis, L Kremer for anti-Dido antibody, the CNB Animal Facility for mouse handling and C Mark for editorial assistance. This work was supported in part by grants from the Spanish Ministry of Science and Innovation (SAF2010-21205 and PIB2010BZ-00564), the Madrid regional government (CAM S-BIO-0189-2006) and the EU (LSHB.CT-2005-518167). The Department of Immunology and Oncology was founded and is supported by the Spanish National Research Council (CSIC) and by Pfizer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by R De Maria
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Fütterer, A., Raya, Á., Llorente, M. et al. Ablation of Dido3 compromises lineage commitment of stem cells in vitro and during early embryonic development. Cell Death Differ 19, 132–143 (2012). https://doi.org/10.1038/cdd.2011.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.62
Keywords
This article is cited by
-
The SPOC proteins DIDO3 and PHF3 co-regulate gene expression and neuronal differentiation
Nature Communications (2023)
-
Hepatitis, testicular degeneration, and ataxia in DIDO3-deficient mice with altered mRNA processing
Cell & Bioscience (2022)
-
Role of DIDO1 in Progression of Esophageal Squamous Cell Carcinoma
Journal of Gastrointestinal Cancer (2020)
-
Dido3-dependent HDAC6 targeting controls cilium size
Nature Communications (2014)
