
Abstract
We propose that the apoptotic function of p53 has an important role in B-cell homeostasis, which is important for the prevention of B-cell lymphomas. We created a mouse model (mΔpro) that lacked residues 58–88 of the proline-rich domain of p53. mΔpro is defective for apoptosis, but is able to arrest cell-cycle progression in hematopoietic tissues. mΔpro develops late-onset B-cell lymphoma, but not the thymic T-cell tumors found in p53-null mice. Interestingly, mΔpro lymphomas comprised incorrectly differentiated B cells. B-cell irregularities were also detected in mΔpro before tumor onset, in which aged mice showed an increased population of inappropriately differentiated B cells in the bone marrow and spleen. We predict that by keeping B-cell populations in check, p53-dependent apoptosis prevents irregular B cells from eventuating in lymphomas.
Similar content being viewed by others

Main
Mutations in TP53 that either inactivate or alter the normal function of p53 are commonplace in human cancer (www-p53.iarc.fr). In hematological malignancies, TP53 mutations are associated with a poorer prognosis.1 This is true for the most common types of non-Hodgkin's lymphomas, diffuse large B-cell lymphoma (DLBCL), and follicular lymphoma, in which TP53 mutations are associated with poorer overall survival and increased tumor aggression.2, 3, 4 Mice with a defective p53 function are also susceptible to lymphoma. Mice deleted for the mouse p53 gene (Trp53) rapidly develop lymphocytic tumors of the thymus,5, 6 and those harboring certain mutations in Trp53 are also prone to T- or B-cell lymphomas.7, 8, 9
How p53 functions to prevent cancer, or specifically lymphoma, is not entirely clear. p53 is a transcription factor that regulates cell survival and proliferation, and is ‘activated’ in response to many different types of cellular stress.10 After DNA damage, p53 has an important role in promoting DNA repair and inducing apoptosis of cells, as well as in causing transient and permanent cell-cycle arrest (senescence). Which of these many activities are responsible for tumor suppression activity is unclear, but the favorites are apoptosis and senescence.11, 12
The proline-rich domain (PRD; residues 62–91) of p53 has been reported to be necessary for the induction of apoptosis.13, 14, 15, 16, 17 However, transfection experiments with different PRD deletion mutants led us to conclude that the PRD is largely dispensable for apoptosis.16 Rather than being the apoptotic domain, we suggested that PRD had a structural role that influenced multiple p53 functions. Nevertheless, one of our PRD mutants (Δ58–88) was markedly defective in apoptosis. The removal of residues 58–88 essentially corresponds to the PRD-deficient human p53, as described by Walker and Levine.13 We created a mouse model for the Δ58–88 mutant, referred to as mΔpro, and describe the findings in this study. Three other proapoptotic PRD mutant mouse models have been reported. The p53Δp mouse had PRD residues 75–91 deleted. This mutant retained a proapoptotic response, although compromised, but was defective for a cell-cycle arrest response.18 The two other PRD mutants, one of which disabled the PXXP motifs, converting the proline residues to alanines (p53AXXA), and the other in which Pin 1 isomerization sites were altered (p53TTAA), showed normal apoptotic responses.19 Thus, the importance of PRD in apoptosis control and tumor suppression remains unresolved.
Our results show that mΔpro is defective for apoptosis in hematopoietic tissues both in vivo and in vitro after DNA damage, but can induce an arrest of cell-cycle progression. Thus, PRD is important for apoptosis to occur. mΔpro mice develop late-onset lymphomas comprising incorrectly differentiated B cells, but are protected against thymic T-cell tumors. mΔpro mice also show B-cell irregularities before tumor onset. mΔpro mice, aged (7 months old) but not young (5 weeks old), had a greater percentage of less-differentiated B cells in the bone marrow and spleen compared with wild-type mice. Our findings emphasize an important role for p53-mediated apoptosis in maintaining correct B-cell cellularity, and in the prevention of B-cell tumors.
Results
Characterization of mΔpro
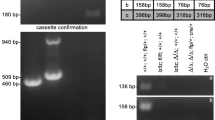
A mutant Trp53 mouse was constructed in which amino acid residues 58–88 were deleted (mΔpro, Figure 1a), as described in the Materials and Methods section. The mΔpro allele is transcribed as determined by reverse-transcriptase PCR (Figure 1b), showing a smaller PCR fragment as expected. Approximately equal amounts of transcription to the wild-type allele were observed in mΔpro homozygote animals, although the mΔpro allele was expressed slightly less well in heterozygote animals compared with the wild-type allele. This is probably because of differences in PCR efficiencies. mΔpro is translated to levels approximately similar to those of wild-type p53. mΔpro was also stabilized after γ-irradiation and was phosphorylated on serine 18 (Figure 1c), consistent with mΔpro being functionally activated. The results for thymus-derived T cells are shown here, but similar levels of protein expression were observed in splenocytes, spleen-derived B cells, and embryonic fibroblasts (data not shown). Mendelian inheritance of the mΔpro allele was also observed over many matings (data not shown).

Characterization of mΔpro mice. (a) Schematic diagram of the construction of mΔpro mice that produce p53 without amino acids 58–88 inclusive, because of a deletion in exon 4 of mouse Trp53 as outlined in the Materials and Methods section. p53 domains: TAD, transactivation; PRD, proline-rich; 4D, tetramerization; CT, C-terminal regulatory. (b) Reverse transcriptase PCR showing the presence of two mRNA species (298bp and 205bp) in thymocyte RNA from the mΔpro heterozygote (p53+/ mΔpro), and a single species in wild-type (p53+/+) (298bp) and mΔpro (205bp) genotypes. No signal was detected in RNA from p53−/−cells, or in the no-template control. (c) Examples of protein expression for each genotype using western blotting from extracts of untreated or γ-irradiated (4Gy) thymocytes. Protein extracts were prepared 2 and 5 h after γ-irradiation and western blotting was carried out to detect p53 and p53 phosphoserine 18. mΔpro protein is translated, with no signal detected in protein extracts from p53−/− cells. β-actin was used as a loading control
mΔpro is defective for DNA damage-induced apoptosis
To address whether mΔpro is defective in apoptosis after DNA damage, we examined the ability of mΔpro to induce apoptosis in vitro and in vivo in several hematopoietic tissues after DNA damage. Thus, 4 Gy of γ-irradiation and 1 μg/ml of the topoisomerase 2 inhibitor, amsacrine,20 elicited strictly p53-dependent apoptosis as observed by comparing Annexin-V staining of thymocytes from p53+/+ and p53−/− mice after DNA damage. Thymocytes exposed to the optimized doses of DNA-damaging agents (Figure 2a) showed a sixfold increase in apoptosis above untreated control levels for p53+/+ cells with both agents, and in cells from mice heterozygous for wild-type p53 and mΔpro (p53+/mΔpro). Little apoptosis, above the levels in untreated controls, was observed in mΔpro and p53−/− cells. The fact that B cells also undergo apoptosis in a p53-dependent manner was confirmed by examining CD45R+ (B220) cells isolated from bone marrow and treated with amsacrine (0.2 μg/ml). Apoptotic cells were labeled with the active caspase probe, FLICA-FAM-VAD-FMK probe (Immunochemistry Technologies, Bloomington, MN, USA), and apoptosis was measured by detecting the FAM-conjugated fluorochrome using flow cytometry. B cells from p53+/+ mice showed a fivefold increase in apoptosis after DNA damage, but no apoptosis above untreated controls was observed for mΔpro or p53−/− cells (Figure 2b).

mΔpro is consistently impaired for apoptosis after DNA damage. In vitro apoptosis analyses. (a) Thymocytes from mice with genotypes p53+/+, mΔpro, heterozygous p53+/mΔpro, and p53−/− were left untreated (U), or treated (T) with a single dose of 4 Gy of γ-irradiation or 1 μg/ml of amsacrine. At 6 h after treatment, apoptosis was measured using the apoptotic marker Annexin-V and PI staining, followed by flow cytometry. (b) B cells (CD45R+) sorted from bone marrow cells from p53+/+, mΔpro, and p53−/− mice were left untreated (U), or were treated (T) with 0.2 μg/ml of amsacrine. At 5 h after treatment, apoptosis was measured using FLICA poly-caspase probe (Immunochemistry Technologies) staining and flow cytometry. In vivo apoptosis analysis. (c) p53+/+, mΔpro, and p53−/− mice were left untreated (U), or were treated (T) with a single dose of 4 Gy of γ-irradiation at 5–6 weeks of age. At 8 h after treatment, mice were administered the FLIVO poly-caspase probe (Immunochemistry Technologies) by tail vein injection, and killed 30 min later. The thymus and spleen were removed and single-cell suspensions of each were analyzed using flow cytometry. All results: mean ±1 S.D. from three separate experiments. Detection of apoptotic cells in thymocytes using flow cytometry is shown in C (boxed)
For in vivo apoptosis analysis, mice were treated with a single 4 Gy dose of γ-irradiation and various tissues were labeled in vivo with the FLIVO-FAM-VAD-FMK probe, which binds activated caspases.21 Apoptosis was measured by identifying FAM-conjugate positive cells using flow cytometry (Figure 2c). The results show a 13-fold increase in apoptosis above control levels in the thymus and a fourfold increase in the spleen (Figure 2c) for p53+/+ mice, but little apoptosis above the untreated controls was observed for mΔpro or p53−/− mice. In summary, mΔpro is substantially impaired for p53-mediated apoptosis in response to DNA damage.
mΔpro is unable to transactivate proapoptotic genes
p53 has been reported to induce apoptosis after DNA damage by either transactivating proapoptotic genes through interaction with ASPP proteins,22, 23 or by facilitating apoptosis directly at the mitochondrial membrane.24, 25PRD has been reported to be required for both processes. To confirm that p53-dependent transactivation is critical for apoptosis in hematopoietic cells,26, 27 thymocytes were γ-irradiated and treated with Actinomycin D (Act-D) to block transcription. Leptomycin B (Lept-B) was also used to prevent nuclear export of p53. The results (Figure 3a) show that there was a sixfold increase in apoptosis in p53+/+ thymocytes after irradiation that was completely ablated by Act-D but unaffected by Lept-B. These data suggest that p53-dependent transactivation is necessary for apoptosis, whereas export of p53 from the nucleus is not the predominant mechanism.

mΔpro is activated after DNA damage but is unable to transactivate proapoptotic genes. Thymocytes from mice with genotypes p53+/+, mΔpro, heterozygous p53+/mΔpro, and p53−/− were left untreated, or were treated with 4 Gy of γ-irradiation or 1 μg/ml amsacrine. (a) Cell samples were treated with 1.25 μg/ml Act-D or with 40 ng/ml Lept-B; at 6 h after treatment, cells were stained with Annexin-V and PI and were analyzed using flow cytometry. (b) Cells were pulsed with 4 Gy of γ-irradiation, and 2 and 5 h later, cells were harvested, RNA was prepared, and levels of mRNA to Bax and Puma were measured by quantitative real-time PCR. Results (mean ±1 S.D.) are from three separate measurements (a, b), and are expressed as the fold difference between untreated and treated samples at each time point (b)
We analyzed whether mΔpro is able to transactivate appropriate downstream proapoptotic p53-dependent genes. Thymocytes were treated with γ-irradiation and RNA was harvested 2 and 5 h later. Quantitative real-time PCR was used to determine the mRNA levels of apoptotic genes Bax and Puma. p53+/+ induced Bax by approximately threefold and Puma by fivefold by 5 h. For mΔpro cells, the increase in apoptotic gene transcription was attenuated with no increase at 2 h, and delayed, with a twofold increase in Bax and Puma by 5 h (Figure 3b). No increase was found in extracts of p53−/− cells. Thus, the failure to induce apoptosis after DNA damage might be explained by the inability of mΔpro to transactivate proapoptotic genes efficiently.
mΔpro causes cell-cycle arrest in bone marrow cells after DNA damage
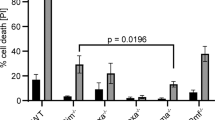
In vitro studies have indicated that deletion of PRD impairs apoptosis, but not the ability of p53 to induce cell-cycle arrest.15, 28, 29, 30 However, p53ΔP mice were proapoptotic but defective for cell-cycle arrest.18 To test whether mΔpro is able to induce cell-cycle arrest, mice were irradiated and at indicated times pulsed with BrdU. Various tissues were harvested, stained with the DNA fluorochrome 7-AAD and an anti-BrdU antibody, and analyzed using flow cytometry. Very little BrdU was found to be incorporated in cells harvested from the spleen or thymus of p53+/+, mΔpro, and p53−/− mice, suggesting that few cycling cells exist in these tissues (data not shown). However, BrdU incorporation was detected in bone marrow. As expected, a reduced S-phase (BrdU positive) population was observed in irradiated p53+/+ cells compared with untreated cells. BrdU-positive populations for all genotypes from untreated and irradiated animals were determined and are shown graphically in Figures 4a and b. By 8 h after irradiation, BrdU incorporation was reduced by approximately threefold in cells from p53+/+ mice, with a 30% reduction in the S phase in cells from mΔpro mice and 15% in p53−/− mice. The difference in the proportion of S-phase cells between mΔpro and p53−/− mice did not reach significance (P=0.055). However, by 24 h after irradiation, BrdU incorporation was reduced >twofold in both p53+/+ and mΔpro mice, and in p53+/mΔpro heterozygotes, but no reduction was observed in p53−/− mice (Figure 4b). At 24 h, the difference between mΔpro and p53−/− mice was very significant (P<0.0001). Thus, mΔpro is capable of restricting cell-cycle progression after DNA damage, although it is less effective than p53+/+.

mΔpro causes cell-cycle slowing of bone marrow cells after DNA damage. p53+/+, mΔpro, p53−/−, and mice heterozygous for mΔpro (p53+/mΔpro and p53−/mΔpro) were left untreated or given a single dose of 4 Gy of γ-irradiation at 5–6 weeks of age. At 8 h (a) or 24 h (b) after treatment, mice were administered 1.5 mg of BrdU by intraperitoneal injection, and killed 90 min later. Bone marrow was removed, and the S-phase cell population was distinguished using flow cytometry, as shown in (b) (boxed). Results: mean ± 1 S.D. from three separate experiments
mΔpro mice are resistant to thymic tumors but prone to B-cell lymphoma
As apoptosis is thought to be necessary for the prevention of thymic tumors, the most common spontaneous tumor observed in p53−/− mice, we asked whether mΔpro mice are susceptible to tumorigenesis. Cohorts of p53+/+, p53−/−, mΔpro, and heterozygous p53+/mΔpro and p53−/mΔpro mice were monitored over time for spontaneous tumor development. In addition, p53+/+, p53+/mΔpro, and mΔpro mice were exposed to 2 Gy of γ-irradiation and were also monitored over time. The results for untreated mice (Figure 5a) show that, over 600 days, p53+/+ and p53+/mΔpro mice are completely tumor resistant, whereas all p53−/− mice had been killed because of tumor burden by 250 days, with a median survival of 144 days. The bulk of p53−/− mice died of thymus-derived T-cell tumors, consistent with previous reports.5, 6 By contrast, mΔpro mice had a longer survival period, with the first tumor-burdened mouse killed at day 350. The mΔpro cohort had a median survival time of 423 days, with the entire cohort dead from tumors by day 550. Necropsies and histopathological analyses of affected animals revealed that most animals died of B-cell lymphoma (Table 1), involving at least one lymph node (predominantly the mesenteric) and other organs such as the spleen, liver, lungs, and kidneys. A few mice were also found to have sarcomas (fibrosarcomas (n=1) and rhabdomyosarcomas (n=2)). A second cohort of 20 mΔpro mice showed a similar tumor spectrum, with two cases of hemangiosarcomas also identified. Mice heterozygous for mΔpro and p53− (p53−/mΔpro) died with similar kinetics to p53−/− mice, with all but two p53−/mΔpro mice developing thymic T-cell lymphomas. One of the two p53−/mΔpro mice without thymic T-cell lymphoma developed B-cell lymphoma and the second mouse developed osteosarcoma.

mΔpro mice have a reduced lifespan. (a) Survival of untreated mΔpro mice. Kaplan–Meier analysis of tumor-free survival in p53+/+, mΔpro, and p53−/− mice is shown. Mice heterozygous for the mΔpro mutation (p53+/mΔpro and p53−/mΔpro) were also monitored for 600 days. Animals were killed when tumor burden was apparent. (b) Decreased survival of mΔpro mice after DNA damage. Kaplan–Meier analysis of p53+/+, p53+/mΔpro, and mΔpro mice treated with a single dose of 2 Gy of γ-irradiation at 5–6 weeks of age. Mice were monitored for 360 days, and killed when tumor burden was apparent, or at day 361
The results for γ-irradiated mice (Figure 5b) show that, over 360 days, p53+/+ mice are completely tumor resistant, whereas mΔpro mice were all dead by day 270, with a median survival of 220 days. The bulk of these mice died of B-cell lymphoma (Table 1). Predominantly, numerous lymph nodes (particularly the mesenteric, axillary, and brachial nodes) and other organs such as the spleen, liver, lungs, kidneys, and, in one case, the brain were affected. One animal developed a fibrosarcoma. p53+/mΔpro mice were largely tumor resistant after DNA damage; however, four (22%) animals did develop tumors (two mice had B-cell lymphoma and two had fibrosarcoma).
Characterization of tumors from mΔpro mice
An examination of spontaneous and irradiated mΔpro tumors on the basis of histological criteria placed most tumors in the late differentiated B-cell lineage (Table 1). The predominant B-cell malignancy was follicular lymphoma (n=8, 40%) for the spontaneous cohort and diffuse large B-cell lymphoma for the irradiated cohort (n=8, 32%).
Tumor DNA from mΔpro tumors was sequenced to determine whether additional p53 mutations were acquired during tumorigenesis. No additional mutations were identified in Trp53 exon and intron–exon boundaries in the 10 lymphomas (five from the irradiated and five from the spontaneous cohort) sequenced. This suggests that mΔpro tumors are caused because of the impaired function of mΔpro and not by additional p53 mutations.
To further stratify the tumors observed in mΔpro mice, expression of B and T-cell differentiation antigens was measured using flow cytometry. B-cell markers included the early pre-B-cell antigen CD117 (c-kit) and the B-cell antigen CD45R (B220), CD19, and κ-light chain.31 T-cell antigens, CD4 and CD8, were also examined. Examples of CD45R, CD19, and κ-light chain staining on B-lymphoma cells are shown in Figure 6a. The results (Figure 6b) show that all spontaneous tumors in mΔpro mice were positive for CD45R and κ-light chain, and negative for CD117. The tumors were essentially negative for T-cell markers. Thus, the tumors are of B-cell origin. However, one tumor contained >40% of the CD8-positive cells, <5% of the CD4-positive cells, and 25% of the κ-light chain-expressing cells, suggesting that this tumor is likely to be of T-cell origin.

Tumors in mΔpro mice are derived from incorrectly differentiated B cells. Tumors were harvested from mice, single-cell suspensions were prepared, and cells were labeled with monoclonal antibodies to seven different B- and T-cell antigenic markers. PE and APC fluorochrome labels were detected using flow cytometry. (a) Examples of flow cytometry for CD45R, CD19, and κ-light chain staining, and the corresponding isotype control (left) on tumor cells. (b) Quantitation of marker expression on tumors derived from mΔpro mice from the spontaneous and DNA-damaged tumor cohorts
γ-irradiated mice stained positive for CD45R and κ-light chain, but were negative for CD117. In this cohort, 4 out of 14 (29%) tumors were positive for CD8 (30–80% of cells) and negative for CD4. By contrast, most tumors from p53−/− and heterozygous p53−/mΔpro mice stained positive for CD8 and rarely with B-cell markers (data not shown). We conclude that mΔpro largely predisposes to B-cell and not T-cell tumors. The absence of CD117 expression suggests that tumors do not derive from early B-cell progenitors.
Atypically, the tumors were largely negative for CD19, which is unexpected in B cells positive for κ-light chain, as this is normally expressed after CD19. To further establish the B-cell immunophenotype of tumors, lymphomas from eight irradiated mice and six lymphomas arising spontaneously were stained with markers to additional B-cell antigens Ly-51 (expressed from the late-pro-B to the small-pre-B stage of B-cell development), CD40 (TNF-receptor family member involved in B-cell activation, and is expressed at stages similar to CD19,32) IgM, and CD22.2 (both markers of late B-cell development and expressed after Ly-51). All spontaneous cohort tumors stained positive for IgM, but were negative for CD40, CD22.2, and Ly51 (data not shown). The same result was found for the irradiated tumor cohort, with one exception (a tumor negative for IgM, CD22.2, and Ly51). Finally, to be sure all markers were present on the same population of tumor cells, multicolor flow cytometry was carried out on tumors from four animals. Results showed the tumors to be positive for CD45R, IgM, and κ-light chain, and negative for CD19 (data not shown). Data for IgM and Ly51 staining are consistent with the tumor histopathological classification of mature B cells, but, similar to CD19, the CD40 and CD22.2 negative results are inconsistent with a mature B-cell phenotype.
Many B-cell markers, such as CD19 and CD22, are downregulated during differentiation of mature B cells to plasma cells.33 To analyze whether the tumors are of plasma cell origin (plasmacytomas), sera from four mice in the spontaneous and eight mice from the irradiated cohort, collected at necropsy, were subjected to agarose gel electrophoresis. No peak in serum immunoglobulins was apparent on total protein staining and on comparison with sera from age-matched p53+/+ mice, suggesting that the tumors are not plasmacytomas (data not shown). Together, these results suggest that mΔpro B-cell tumors arose late in B-cell development from incorrectly differentiated B cells, and are unlikely to have been derived from plasma cells.
Characterization of B cells in aged mΔpro mice
If the proline domain of p53, and hence p53-directed apoptosis, is required to remove incorrectly differentiated B cells, it could be expected that mΔpro animals would accumulate incorrectly differentiated B cells before tumor onset. To test this hypothesis, bone marrow cells and splenocytes were removed from young mΔpro mice (5 weeks old) and aged mice (7 months old), as well as from age-matched p53+/+ controls, and were stained for B-cell markers CD45R, CD19, and κ-light chain. At 5 weeks of age, no differences were significant between the mΔpro and p53+/+ genotypes in the percentage of CD45R+ CD19− and of CD45R+ CD19+ cells in the bone marrow and spleen (Figure 7a). However, aged mΔpro mice had a higher percentage of CD45R+ CD19− cells (P=0.0004) and a lower percentage of CD45R+ CD19+ cells (P<0.0001) in the bone marrow (Figure 7b). A similar result was found for the spleen, with the exception of one animal that had a CD45R+ CD19+ immunophenotype similar to that of p53+/+ animals (Figure 7b). Although aged mΔpro had more of the less-differentiated (CD45R+ CD19−) cells, they did not have the same immunophenotype as that of mΔpro tumor cells on the basis of κ-light chain staining (Figure 7b). This result was confirmed by triple staining bone marrow and splenocytes isolated from a second cohort of 7-month-old mΔpro and p53+/+ animals with the CD45R, CD19, and κ-light chain antigens (boxed insert Figure 7b). These data suggest that without the apoptotic function of p53, fewer properly differentiated B cells accumulate in the bone marrow and spleen, which becomes more apparent with increasing age.

Aged, but not young, mΔpro mice have increased populations of early pro-B cells. Bone marrow cells and splenocytes were harvested from mΔpro and p53+/+ mice at 5 weeks or 7 months of age. Single-cell suspensions were labeled with monoclonal antibodies to three B-cell antigenic markers: CD45R, CD19, and κ-light chain. (a) Quantitation of marker expression on cells derived from 5-week-old mice. (b) Quantitation of marker expression on cells derived from 7-month-old mice. The results are from six mice per genotype and from three independent experiments. Boxed insert: triple staining using the CD45R, CD19, and κ-light chain markers on cell suspensions from a second cohort of 7-month-old mice
Discussion
This paper describes the characterization of mutant p53 mice, deleted for residues 58–88, which include the entire PRD (mΔpro). mΔpro was found to be expressed at similar levels to wild-type p53, is stabilized after DNA damage, and is phosphorylated on serine 18. However, mΔpro is defective for the induction of apoptosis after DNA damage in hematopoietic cells in vitro and in vivo. As transactivation seems to be required for p53-mediated apoptosis to occur (Figure 3a),26, 30, 34, 35, 36 and does not seem to require the export of p53 to mitochondria, the poor ability of mΔpro to transactivate proapoptotic genes (at best twofold) provides a possible explanation for this defect.13, 14, 15, 24, 28, 29, 30 In contrast, mΔpro is able to cause a slowing of cell-cycle progression (Figure 4), although less well than wild-type p53. These results are consistent with PRD being necessary for the efficient induction of apoptosis by p53.
The previously described PRD mutant mouse p53ΔP showed some similarities and differences to mΔpro.18 p53ΔP was markedly defective for transactivation and cell-cycle arrest, whereas mΔpro was less impaired for these functions. These differences might be best explained by the low-level expression of p53ΔP, as it is very sensitive to Mdm2-dependent proteolysis, whereas mΔpro is not. The differences in Mdm2 sensitivity might be due to the slightly altered protein structures caused by deletions, as has been suggested previously.16, 19 However, treatment of p53ΔP/E1A-expressing MEFs with high doses of DNA-damaging agent adriamycin did elicit an apoptotic response, whereas mpro is defective for apoptosis in hematopoietic cells at relatively modest levels of DNA damage. It is therefore possible that mΔpro retains some apoptotic ability if higher doses of DNA-damaging agents or nonhematopoietic tissues are used.
Despite its defects, the p53ΔPmutant mouse could prevent the formation of spontaneous thymic lymphomas, but as it was not apoptosis defective, the importance of apoptosis for the suppression of thymic tumors was not resolved.18 We examined the ability of mΔpro to suppress spontaneous tumors over 600 days and γ-irradiation-induced tumors over 1 year. In both cases, mΔpro, similar to p53−P, is largely able to suppress the formation of thymic tumors, although these were commonplace in p53-null mice. As mΔpro is unable to induce apoptosis in thymic cells in our experiments, but is able to suppress thymic tumors, it would seem that p53-dependent apoptosis is not required for the suppression of such tumors. However, as there are reports suggesting otherwise,37, 38, 39, 40 we conclude that apoptosis is not always required for the prevention of thymic tumors. Furthermore, as mice deleted for the gene encoding p21Waf1/Cip1 are not prone to thymic tumors,41 it seems unlikely that the induction of cell-cycle arrest by p53 prevents thymic tumors. Thus, another mechanism might be required, such as activation of DNA repair pathways.
mΔpro mice are predisposed to late-onset B-cell lymphomas. Follicular B-cell lymphoma was the predominant tumor that developed spontaneously in mΔpro mice, with the more aggressive, diffuse B-cell lymphoma being the dominant tumor type after administration of γ-irradiation (Table 1). Few T-cell tumors developed in mΔpro tumor cohorts. On the basis of morphology and the CD45R+, IgM+, κ-light chain+, and CD117− immunophenotype, tumors were classified as mature B cells. As mΔpro is defective for apoptosis after DNA damage in B cells, it seems likely that p53-dependent apoptosis is the necessary activity for the prevention of B-cell tumors. Other apoptotic-defective mouse models also develop B-cell lymphomas, including mice with S23A or R172P mutations in p53;8, 9 mice lacking both proapoptotic genes Bim and Puma;42 and mice deficient for the proapoptotic protein, Bad.43 The role of p53 in inducing prolonged cell-cycle arrest after DNA damage has been proposed to be important for tumor suppression.12, 44 mΔpro allows cell-cycle slowing in bone marrow, similar to p53+/mΔpro heterozygotes, which are not tumor prone. It would seem therefore that cell-cycle arrest may not be very important for the suppression of B-cell lymphoma. However, the arrest in cell-cycle progression was delayed and attenuated in mΔpro compared with p53+/+ cells and this may assist tumor development.
Of considerable interest is our finding of incorrectly differentiated cells in mΔpro B-cell tumors. mΔpro B-cell tumors were incorrectly differentiated for B-cell antigens CD19, CD40, and CD22.2. The lack of CD19 and CD40 expression was unusual as the tumor cells did express the later B-cell development markers, IgM and κ-light chain. The lack of CD22.2 expression was also inconsistent with the histopathological examination that placed tumors into classifications of largely follicular or diffuse large B-cell lymphomas, as normally mature B-cell malignancies are expected to express CD22.2. The existence of plasmacytomas, which contain plasma cells that lose the expression of B-cell markers during differentiation from mature B cells, is unlikely, owing to the absence of increased serum immunoglobulins in mΔpro tumor-burdened mice, although without further analysis it cannot be formally excluded at this stage, as occasional human plasmacytomas do present without high serum immunoglobulin. The absence of CD19 impairs B-cell maturation, as is evident from CD19-null mice.45 CD19-deficient mice were also associated with increased B-cell receptor editing to compensate for a defective positive selection. However, CD19-deficient mice are mostly normal,46, 47 and it is not known whether increased B-cell receptor editing, associated with CD19-negative B cells, augments the tumor susceptibility on a p53-defective background such as mΔpro. This is the first report of incorrectly differentiated B-cell tumors associated with p53 mutations in mouse models.
Although, p53 mutations are very prominent in some cancer types, they are not uncommon in human lymphomas, occurring in 15–20% of cases, and are often associated with poorer prognosis.2, 3, 4, 48 In addition, individuals with germline p53 mutations (Li-Fraumeni syndrome) develop B-cell lymphomas.49, 50, 51, 52 CD19-negative cases of B-cell lymphoma exist, accounting for 6–14% of cases, and a weak expression of CD19 is prevalent in follicular B-cell lymphoma (50–70%) and diffuse large B-cell lymphoma (24–50%).53, 54, 55, 56 Of particular interest, on the basis of the findings of this study, is to determine whether CD19-negative cases of human B-cell lymphomas are associated with p53 mutations.
Before tumor development, mΔpro animals show an increase in altered B-cell cellularity in the bone marrow and spleen. Seven-month-old mΔpro mice had a significant increase in the number of early pro-B cells (CD45R+ CD19−) and a significant reduction in B cells later in development (CD45R+ CD19+), compared with p53+/+ mice. A similar finding was found for mice lacking both proapoptotic genes Bim and Puma,42 and for mice lacking the micro-RNA miR-17–92 that represses Bim.57 Erlacher et al. (2006) showed an increased percentage of less-differentiated cells in the bone marrow in apoptotic-defective mice, and according to Ventura et al. (2008), by repressing Bim, miR-17–92 promotes the survival of B-cell progenitors, although the CD19 phenotype was not reported. These results are consistent with our finding that without the apoptotic function of p53, a pool of very early B cells accumulates in bone marrow with age.
In summary, deletion of the PRD in mouse p53 (mΔpro) prevents DNA damage-induced apoptosis in hematopoietic tissues, which is likely to be because of a reduced ability to transactivate proapoptotic genes. mΔpro mice are prone to B-cell lymphomas but not to T-cell tumors, which are characteristic of p53−/− mice. mΔpro lymphomas comprise late incorrectly differentiated B cells. Exposure to γ-irradiation resulted in a more aggressive B-cell tumor type. Before tumor onset, aged mΔpro animals accumulate progenitor B cells in the bone marrow and spleen. We suggest that p53-dependent apoptosis is required for B-cell homeostasis, and predict that p53 may have a role in B-cell differentiation in vivo. Together, these p53 functions prevent the accumulation of progenitor B cells, thereby reducing the likelihood of incorrectly differentiated B cells persisting that can subsequently develop into lymphomas.
Materials and Methods
Generation of mΔpro mice
mΔpro mice were developed by Ozgene (WA, Australia). The targeting vector used a genomic fragment of Trp53 that included introns 1–10, and is detailed in Figure 1a. The neomycin-resistance gene was flanked by FRT signal sequences. The mΔpro mutation, a 93 bp deletion in exon 4, was introduced into the vector by overlap extension PCR. The targeting vector was introduced into C57BL/6 ES cells. Correct gene targeting was confirmed using a panel of PCRs (the primer sequences are available by request). All coding and intron–exon boundaries were sequenced to ensure that the correct mutation and no other p53 mutations were introduced. Neomycin-resistant, correctly targeted, ES clones were injected into blastocysts. Germline offspring were developed from two independent ES cell clones. Mice were genotyped for mΔpro by PCR. To confirm that the deletion in exon 4 did not affect intron–exon splicing, mRNA was extracted from mΔpro thymocytes, cDNA was prepared, and the Trp53 transcript was amplified by PCR and sequenced (data not shown). The Trp53mΔpro sequence was found to be correct. After breeding to homozygosity, the neomycin-resistance cassette was excised by crossing mΔpro mice with FLPe mice expressing the flippase 1 recombinase. Loss of the neomycin cassette was determined by PCR.
All p53+/+, p53−/−, and FLPe mice were on the C57BL/6 background. FLPe and p53−/− mice were purchased from the Jackson Laboratory, Bar Harbor, ME, USA and were genotyped using PCR (primer sequences are available on request). All mice were housed under SPF conditions, and the approval of the local research ethics committee was obtained before study initiation.
In vitro apoptosis analyses
Thymocytes and bone marrow cells were isolated from 5- to 6-week-old mice, and cultured at 2 × 106 cells/ml in DMEM supplemented with 20% fetal calf serum, L-glutamine (2 mM), and antibiotics. Thymocytes were left untreated, irradiated with a pulse of 4 Gy of γ-irradiation, or treated with 1 μg/ml amsacrine (Sigma-Aldrich, St Louis, MO, USA). Bone marrow cells were left untreated or were treated with 0.2 μg/ml amsacrine. For the apoptosis assay using thymocytes, 1x105 cells were doubled stained with Annexin-V:APC and PI (BD Biosciences, San Jose, CA, USA), and analyzed using flow cytometry. For the apoptosis assay using bone marrow cells, 1 × 106 cells were stained with the FLICA-FAM-VAD-FMK probe (Immunochemistry Technologies), and analyzed using flow cytometry.
In vivo apoptosis analysis
Mice were irradiated with a pulse of 4 Gy γ-irradiation at 5–6 weeks of age. For detection of active caspases, mice were administered the FLIVO-FAM-VAD-FMK probe intravenously into the tail vein (Immunochemistry Technologies) 8 h after irradiation. Mice were killed 30 min later and their thymus and spleen were removed. Single-cell suspensions were analyzed using flow cytometry.
In vivo cell-cycle arrest analysis
Mice were irradiated with a pulse of 4 Gy of γ-irradiation at 5–6 weeks of age. For the detection of cells actively synthesizing DNA, mice were administered 1.5 mg of BrdU (BD Biosciences) by intraperitoneal injection, 8 or 24 h after irradiation. Mice were killed 90 min later, and bone marrow was removed. Incorporated BrdU was stained with the anti-BrdU/APC antibody and total DNA labeled with 7-AAD, and was analyzed using flow cytometry (BD Biosciences).
Protein analysis
Cells were isolated from 4- to 6-week-old animals, pulsed with 4 Gy of γ-irradiation, and placed in complete DMEM for indicated times. Protein lysate concentrations were determined using the BCA assay (Pierce, Rockford, IL, USA), and approximately 20 μg of protein was separated on 10% SDS-PAGE gels (Invitrogen, Carlsbad, CA, USA) for western blotting. Blots were probed with primary antibodies against p53 (1C12, Cell Signaling, Boston, MA, USA), p53 phospho-Ser18 (polyclonal, Cell Signaling), and β-actin (AC-15, Abcam, Cambridge, MA, USA). Alkaline phosphate-conjugated antibodies were detected using the Western Breeze Immunodetection Kit (Invitrogen).
Gene expression analysis
Total RNA was extracted, DNase was treated, precipitated with linear acrylamide (Ambion, Austin, TX, USA), purified (GE Healthcare, Fairfield, CT, USA), and integrity and purity were checked. cDNA was synthesized using oligo-dT primers and MMLV reverse transcriptase (Promega, Madison, WI, USA). For reverse transcriptase PCR (Figure 1b), primer sequences are available on request. Quantitative real-time PCR reactions were performed using the Rotor-Gene 6000 system (Corbett Life Science, Sydney, Australia) with the QuantiTect SYBR Green PCR master mix (Qiagen, Hilden, Germany). Primer sequences for detecting Bax and Puma were, Bax: forward, 5′-TGGCTGGGGAGACACCTG-3′, reverse, 5′-CTCCATATT GCTGTCCAGTTCATC-3′; Puma: forward, 5′-CAAGAAGAGCAGCATCGACA-3′, reverse, 5′-CTCCAGGATCCCTGGGTAAG-3′. The CT method was used for the comparative quantification of gene expression, with results from each time point normalized against β2-M expression. Primer sequences for β2-M were forward, 5′-GCTATCCAGAAAACCCCTCA-3′; reverse, 5′-CGGGTGGAACTGTGTTACG-3′.
Spontaneous tumor analysis
Mice were aged and killed when visible tumor burden was apparent: tumors reached 1 cm in diameter, mice lost more than 20% of their adult body weight, or developed a hunched posture in the case of thymic lymphomas. Tissues and tumors were fixed in 10% neutral-buffered formalin and paraffin embedded for histological analysis to identify tumor type on the basis of cellular morphology. On dissection, a part of each tumor was removed, single-cell suspensions were prepared, and cells were analyzed for cell-surface marker expression using flow cytometry (see below).
DNA damage tumor analysis
Mice were irradiated with a single dose of 2 Gy of γ-irradiation at 5–6 weeks of age. Mice were monitored daily and killed when visible tumor burden was apparent (criteria outlined above), or at the end of the study (day 361). Tissues and tumors were processed as described above.
Sequencing Trp53 in Tumor DNA
DNA was extracted from 10 paraffin-embedded lymphomas (five from the irradiated and five from the spontaneous cohort) using the QlAamp DNA micro kit (Qiagen). Tumor DNA was used as the template in PCRs that amplified each Trp53 exon and intron–exon boundaries (primer sequences are available on request). Purified PCR products were sequenced using an ABI3730 DNA analyzer (Applied Biosystems, Scoresby, Victoria, Australia), and were analyzed for mutations using 4Peaks software (Mekentosj, Amsterdam, The Netherlands).
Cell-surface marker analysis
To characterize T- and B-cell malignancies, single-cell suspension of tumors was prepared by collagenase digestion. Cells, 1 × 106, were labeled with antibodies to a panel of 10 lymphoid cell-surface markers (CD45R/APC; CD4/PE, CD40/PE, CD8/APC, CD19/PE, CD22.2/PE, CD117/PE, κ-light chain/PE; Ly-51/PE, and IgM/PE). To characterize B-cell populations in young and aged mΔpro animals, bone marrow and splenocytes from 5-week-old and 7-month-old mΔpro and wild-type mice were double stained with CD45R/APC and CD19/PE, or single stained with κ-light chain/PE. Triple staining (CD45R/APC, CD19/PerCP-Cy5.5, and κ-light chain/PE) was performed on bone marrow and splenocytes isolated from a second cohort of 7-month-old mΔpro and p53+/+ animals. Antibody incubation, 30 min at 4 °C, was followed by a wash step and flow cytometry (BD Biosciences). All analyses included the appropriate isotype-matched-negative control labeled with the same fluorochrome.
Flow cytometry
For flow cytometry, 20–30 000 cellular events were collected using the FACSCalibur system (BD Biosciences) and data were analyzed with CellQuest software (BD Biosciences). For cell sorting, CD45R+ cells were separated from total bone marrow cells using the FACSAria system (BD Biosciences).
Serum immunoglobulin analysis
Serum proteins from mΔpro mice that developed tumors spontaneously or after irradiation, and that from control p53+/+ animals, were separated using agarose gel electrophoresis and total protein was stained using the TITAN Gel Serum Protein kit (Helena Laboratories, Beaumont, TX, USA).
Statistical analyses
Results are expressed as the mean ±1 S.D. Unless otherwise indicated, results are from at least three independent experiments with samples from at least two p53+/+, two mΔpro, and one p53−/− mice per experiment. Statistical differences between two groups were evaluated using Student's t-test, with P<0.05 taken as a significant difference.
Conflict of Interest
The authors declare no conflict of interest.
Abbreviations
- BrdU:
-
bromo-deoxyuridine
- ES:
-
embryonic stem cells
- Δ, deletion, Pin1:
-
prolyl isomerise 1
- PCR:
-
polymerase chain reaction
- TP53 :
-
human p53 gene
- Trp53 :
-
mouse p53 gene
References
Preudhomme C, Fenaux P . p53 and hematologic malignancies. Pathol Biol (Paris) 1997; 45: 898–908.
O'Shea D, O'Riain C, Taylor C, Waters R, Carlotti E, Macdougall F et al. The presence of TP53 mutation at diagnosis of follicular lymphoma identifies a high-risk group of patients with shortened time to disease progression and poorer overall survival. Blood 2008; 112: 3126–3129.
Young KH, Weisenburger DD, Dave BJ, Smith L, Sanger W, Iqbal J et al. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood 2007; 110: 4396–4405.
Ichikawa A, Hotta T, Takagi N, Tsushita K, Kinoshita T, Nagai H et al. Mutations of p53 gene and their relation to disease progression in B-cell lymphoma. Blood 1992; 79: 2701–2707.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery Jr CA, Butel JS et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356: 215–221.
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994; 4: 1–7.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119: 861–872.
Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 2004; 36: 63–68.
MacPherson D, Kim J, Kim T, Rhee BK, Van Oostrom CT, DiTullio RA et al. Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. EMBO J 2004; 23: 3689–3699.
Vousden KH, Lu X . Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2: 594–604.
Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445: 661–665.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445: 656–660.
Walker KK, Levine AJ . Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci USA 1996; 93: 15335–15340.
Baptiste N, Friedlander P, Chen X, Prives C . The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 2002; 21: 9–21.
Zhu J, Jiang J, Zhou W, Zhu K, Chen X . Differential regulation of cellular target genes by p53 devoid of the PXXP motifs with impaired apoptotic activity. Oncogene 1999; 18: 2149–2155.
Edwards SJ, Hananeia L, Eccles MR, Zhang YF, Braithwaite AW . The proline-rich region of mouse p53 influences transactivation and apoptosis but is largely dispensable for these functions. Oncogene 2003; 22: 4517–4523.
Ruaro EM, Collavin L, Del Sal G, Haffner R, Oren M, Levine AJ et al. A proline-rich motif in p53 is required for transactivation-independent growth arrest as induced by Gas1. Proc Natl Acad Sci USA 1997; 94: 4675–4680.
Toledo F, Krummel KA, Lee CJ, Liu CW, Rodewald LW, Tang M et al. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell 2006; 9: 273–285.
Toledo F, Lee CJ, Krummel KA, Rodewald LW, Liu CW, Wahl GM . Mouse mutants reveal that putative protein interaction sites in the p53 proline-rich domain are dispensable for tumor suppression. Mol Cell Biol 2007; 27: 1425–1432.
Johnson RK, Wodinsky I, Swiniarski J, Meaney KF, Clement JJ . Interaction of gamma-irradiation with two new antineoplastic agents, aziridinylbenzoquinone (AZQ) and 4′- (acridinylamino)methanesulfon-m-anisidide (AMSA), in murine tumors in vivo. Int J Radiat Oncol Biol Phys 1979; 5: 1605–1609.
Griffin RJ, Williams BW, Bischof JC, Olin M, Johnson GL, Lee BW . Use of a fluorescently labeled poly-caspase inhibitor for in vivo detection of apoptosis related to vascular-targeting agent arsenic trioxide for cancer therapy. Technol Cancer Res Treat 2007; 6: 651–654.
Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 2001; 8: 781–794.
Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat Genet 2006; 38: 1133–1141.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303: 1010–1014.
Chipuk JE, Maurer U, Green DR, Schuler M . Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 2003; 4: 371–381.
Attardi LD, Lowe SW, Brugarolas J, Jacks T . Transcriptional activation by p53, but not induction of the p21 gene, is essential for oncogene-mediated apoptosis. EMBO J 1996; 15: 3693–3701.
Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T . p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993; 362: 847–849.
Roth J, Koch P, Contente A, Dobbelstein M . Tumor-derived mutations within the DNA-binding domain of p53 that phenotypically resemble the deletion of the proline-rich domain. Oncogene 2000; 19: 1834–1842.
Sakamuro D, Sabbatini P, White E, Prendergast GC . The polyproline region of p53 is required to activate apoptosis but not growth arrest. Oncogene 1997; 15: 887–898.
Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L, Debussche L . The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J 1998; 17: 4668–4679.
Hardy RR, Kincade PW, Dorshkind K . The protean nature of cells in the B lymphocyte lineage. Immunity 2007; 26: 703–714.
Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A . A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci USA 1992; 89: 6550–6554.
Liu YJ, Banchereau J . Mutant mice without B lymphocyte follicles. J Exp Med 1996; 184: 1207–1211.
Jimenez GS, Nister M, Stommel JM, Beeche M, Barcarse EA, Zhang XQ et al. A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat Genet 2000; 26: 37–43.
Chao C, Saito S, Kang J, Anderson CW, Appella E, Xu Y . p53 transcriptional activity is essential for p53-dependent apoptosis following DNA damage. EMBO J 2000; 19: 4967–4975.
Sabbatini P, Lin J, Levine AJ, White E . Essential role for p53-mediated transcription in E1A-induced apoptosis. Genes Dev 1995; 9: 2184–2192.
Braun FK, Fecker LF, Schwarz C, Walden P, Assaf C, Durkop H et al. Blockade of death receptor-mediated pathways early in the signaling cascade coincides with distinct apoptosis resistance in cutaneous T-cell lymphoma cells. J Invest Dermatol 2007; 127: 2425–2437.
Klemke CD, Brenner D, Weiss EM, Schmidt M, Leverkus M, Gulow K et al. Lack of T-cell receptor-induced signaling is crucial for CD95 ligand up-regulation and protects cutaneous T-cell lymphoma cells from activation-induced cell death. Cancer Res 2009; 69: 4175–4183.
Chen J, Fiskus W, Eaton K, Fernandez P, Wang Y, Rao R et al. Cotreatment with BCL-2 antagonist sensitizes cutaneous T-cell lymphoma to lethal action of HDAC7-Nur77-based mechanism. Blood 2009; 113: 4038–4048.
Kampa KM, Acoba JD, Chen D, Gay J, Lee H, Beemer K et al. Apoptosis-stimulating protein of p53 (ASPP2) heterozygous mice are tumor-prone and have attenuated cellular damage-response thresholds. Proc Natl Acad Sci USA 2009; 106: 4390–4395.
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P . Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82: 675–684.
Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med 2006; 203: 2939–2951.
Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP et al. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci USA 2003; 100: 9324–9329.
Feldser DM, Greider CW . Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 2007; 11: 461–469.
Diamant E, Keren Z, Melamed D . CD19 regulates positive selection and maturation in B lymphopoiesis: lack of CD19 imposes developmental arrest of immature B cells and consequential stimulation of receptor editing. Blood 2005; 105: 3247–3254.
Rickert RC, Rajewsky K, Roes J . Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature 1995; 376: 352–355.
Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF . Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity 1995; 3: 39–50.
Koduru PR, Raju K, Vadmal V, Menezes G, Shah S, Susin M et al. Correlation between mutation in P53, p53 expression, cytogenetics, histologic type, and survival in patients with B-cell non-Hodgkin's lymphoma. Blood 1997; 90: 4078–4091.
Malkin D, Li FP, Strong LC, Fraumeni Jr JF, Nelson CE, Kim DH et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250: 1233–1238.
Li FP, Fraumeni Jr JF . Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 1969; 71: 747–752.
Li FP, Fraumeni Jr JF, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA et al. A cancer family syndrome in twenty-four kindreds. Cancer Res 1988; 48: 5358–5362.
Nichols KE, Malkin D, Garber JE, Fraumeni Jr JF, Li FP . Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomarkers Prev 2001; 10: 83–87.
Kimura M, Yamaguchi M, Nakamura S, Imai H, Ueno S, Ogawa S et al. Clinicopathologic significance of loss of CD19 expression in diffuse large B-cell lymphoma. Int J Hematol 2007; 85: 41–48.
Masir N, Marafioti T, Jones M, Natkunam Y, Rudiger T, Hansmann ML et al. Loss of CD19 expression in B-cell neoplasms. Histopathology 2006; 48: 239–246.
Tedoldi S, Mottok A, Ying J, Paterson JC, Cui Y, Facchetti F et al. Selective loss of B-cell phenotype in lymphocyte predominant Hodgkin lymphoma. J Pathol 2007; 213: 429–440.
Yang W, Agrawal N, Patel J, Edinger A, Osei E, Thut D et al. Diminished expression of CD19 in B-cell lymphomas. Cytometry B Clin Cytom 2005; 63: 28–35.
Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008; 132: 875–886.
Acknowledgements
We are grateful for technical assistance from N Bennett, J North, M Schultz, Z Lateef, B Li, X Tan, and L Wallis. L Hananeia and S Edwards are acknowledged for their contributions to the early phases of this study. I Morison, D Speidel, and H Campbell are thanked for comments on the paper. This work was supported by a grant from the Royal Society of New Zealand Marsden Fund, a Cancer Institute NSW Program Grant, and a Health Research Council of New Zealand project grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Oren
Rights and permissions
About this article
Cite this article
Slatter, T., Ganesan, P., Holzhauer, C. et al. p53-mediated apoptosis prevents the accumulation of progenitor B cells and B-cell tumors. Cell Death Differ 17, 540–550 (2010). https://doi.org/10.1038/cdd.2009.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.136
Keywords
This article is cited by
-
Silencing of Testin expression is a frequent event in spontaneous lymphomas from Trp53-mutant mice
Scientific Reports (2020)
-
∆133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling
Nature Communications (2018)
-
Tumor protein 53 mutations are enriched in diffuse large B-cell lymphoma with irregular CD19 marker expression
Scientific Reports (2017)
-
The proline rich domain of p53 is dispensable for MGMT-dependent DNA repair and cell survival following alkylation damage
Cell Death & Differentiation (2017)
-
Δ122p53, a mouse model of Δ133p53α, enhances the tumor-suppressor activities of an attenuated p53 mutant
Cell Death & Disease (2015)
