
Abstract
Mouse embryonic stem (ES) cells remain pluripotent in vitro when grown in the presence of leukemia inhibitory factor (LIF) cytokine. LIF starvation leads to cell commitment, and part of the ES-derived differentiated cells die by apoptosis together with caspase3-cleavage and p38α activation. Inhibition of p38 activity by chemical compounds (PD169316 and SB203580), along with LIF withdrawal, leads to different outcomes on cell apoptosis, giving the opportunity to study the influence of apoptosis on cell differentiation. By gene profiling studies on ES-derived differentiated cells treated or not with these inhibitors, we have characterized the common and specific set of genes modulated by each inhibitor. We have also identified key genes that might account for their different survival effects. In addition, we have demonstrated that some genes, similarly regulated by both inhibitors (upregulated as Bcl2, Id2, Cd24a or downregulated as Nodal), are bona fide p38α targets involved in neurogenesis and found a correlation with their expression profiles and the onset of neuronal differentiation triggered upon retinoic acid treatment. We also showed, in an embryoid body differentiation protocol, that overexpression of EGFP (enhanced green fluorescent protein)–BCL2 fusion protein and repression of p38α are essential to increase formation of TUJ1-positive neuronal cell networks along with an increase in Map2-expressing cells.
Similar content being viewed by others

Main
Mouse embryonic stem (ES) cells derived from the inner cell mass of blastocysts are expanded and maintained pluripotent in vitro in the presence of LIF (leukemia inhibitory factor), an interleukin-6 family member that displays pleiotropic functions, depending on both cell maturity and cell types.1, 2, 3 These pluripotent cells recapitulate the complete mouse developmental program when injected into fertilized eggs.4, 5 In addition, upon aggregation in the absence of LIF, ES cells formed a three-dimensional ball-like cell structure (embryoid bodies, EBs), mimicking the first steps of post-implantation embryos.6 Differentiation could also be induced on monolayer ES cells grown in the absence of LIF. Within the first 48 h of LIF withdrawal, plated ES cells lose their pluripotency and differentiate into heterogeneous cell populations. At the third day, 30% of the differentiated cells die by apoptosis, concomitantly with a peak of p38α mitogen-activated protein kinase (MAPK) activation in the dead cell fraction.7, 8 When this first wave of apoptosis is blocked by the PD169316 compound, a p38 inhibitor, the expression of differentiation markers is altered, emphasizing the importance of p38 activation and apoptosis to control early steps of cell differentiation.9
Neuronal differentiation is efficiently triggered from EBs or plated ES cells treated with retinoic acid (RA), in the absence of serum.10, 11 Growth factors and/or defined medium-based procedures, allowing a gradual differentiation of EBs to neuronal precursor cells, have also been described (Bouhon et al.,12 Gossrau et al.13 and reference therein). Recently, it has been shown that the p38α MAPK activity, which is decreased upon RA treatment, turned out to be an early key regulator of cell fate decision.14 At different time points of cell differentiation, activation or repression of p38α MAPK is critical respectively for the establishment of myogenic and cardiomyogenic or neuronal lineages.15, 16 The p38 MAPK family is composed of 4 isoforms (α, β, δ, γ) that display various effects on cell differentiation and apoptosis.17, 18 P38 activity is increased in various neurodegenerative diseases, including Alzheimer's and Prion-related syndromes, and proper neurons integrity relies on its tight regulation.19, 20, 21 Many reports, based mainly on the use of p38 inhibitors, have concluded on the involvement of p38 kinases in glucose uptake,22 nucleotide metabolism,23 stimulation of the Mnk kinases24 and chromatin remodelling, through the MSK1 kinase.25 However, the fact that p38 inhibitors have different outcomes on ES-derived differentiated cell apoptosis8 indicates that data obtained with these inhibitors could be due to secondary effects of these chemicals along with a modulation in p38 kinase's activity. Also, the beneficial properties of some p38 inhibitors to cure inflammatory diseases stimulate the interest for a comprehensive characterization of the p38 inhibitor targets.17, 26 Knockout mice models, established for each p38 isoform, revealed the physiologic non-redundant function of the p38α gene in placental organogenesis and erythropoiesis,27, 28 whereas no embryonic defects were reported for the other isoforms (reviewed by Ihle28 and Aouadi et al.29). However, the specific transcriptional targets of p38α that might be involved in early differentiation processes remain unknown.
By combining gene profiling experiments performed with p38 α and β inhibitors with expression studies conducted with a p38α−/− ES cell line, we identified, in addition to specific inhibitor-regulated genes, some bona fide p38α target genes. We noticed that some of these genes are known to regulate apoptosis or neurogenesis and demonstrated that the Bcl2 gene, whose expression level is directly correlated to p38α activation, triggers the formation of neuronal networks when overexpressed in a global p38α-repressed environment.
Results
Identification of SB203580-regulated genes in mouse ES-derived differentiated cells
We have previously demonstrated that the pharmacological p38 inhibitors PD169316 and SB203580, widely used to study the p38 MAPK pathway, have different outcomes on ES-derived differentiated cells. Indeed, although PD169316 has an antiapoptotic effect on ES-derived precursor cells, SB203580, in contrast, has an opposite effect and amplifies the LIF-withdrawal-induced cell death process.8 This increase in apoptosis was also observed in the LIF-deprived p38α−/− ES cell line compared to the WT ES cell line.8
The percentage of apoptotic cells, as measured by flow cytometry, was about 30% at d3 (cells grown for 3 days without LIF) and 50% at d3SB (cells grown for 3 days without LIF in the presence of SB, not shown), whereas a similar level of cleavage of caspase 3 was detected in these apoptotic samples (Figure 1). This indicates that an apoptotic pathway, independent of caspase 3, was activated by the SB compound, leading to an increase in LIF-withdrawal-induced apoptosis of differentiated precursor cells. To identify the common and specific target genes of each inhibitor and to characterize those that are potentially involved in differential apoptosis regulation, we undertook a microarray analysis with the SB compound and compared the results with previous work performed with the PD compound.9

Opposite effects of two p38 inhibitors, PD169316 (PD) and SB203580 (SB), on LIF-withdrawal induced apoptosis in ES-derived differentiated cells. Protein RIPA cell extracts from ES cells, grown for 3 days in the presence (+) or absence of LIF (−), without (−) or with 10 μM of PD or SB, were analyzed by western blot with the antibodies as indicated: caspase 3 (CASP-3) and ERK2 (used as a loading constant control)
Gene profiling experiments were carried out with mRNAs prepared from two different ES cell lines (S1 and D4) grown for 3 days in cell medium without LIF (d3) in the absence or in the presence of 10 μM SB203580 (d3SB). Total RNAs from plated ES cell lines grown under these conditions (d3 or d3SB) were processed and hybridized on the mouse MG_U74A and MG_U74AV2 chips (Affymetrix), which include 10 043 transcripts (Materials and Methods). Pair-wise comparisons were performed with the DMT3 and MAS5 softwares. The mean of the ‘signal’ for each gene in every cell growth condition was calculated. Genes quoted as ‘present’ or ‘absent’ were retrieved with the Affymetrix parameters. For each pair-wise comparison, genes quoted ‘absent’ in both conditions were eliminated. To identify genes differentially expressed between experimental groups, we used the nonparametric Wilcoxon rank sum test (or Mann–Whitney test) as detailed in Materials and Methods. In addition, to identify genes potentially involved in differential apoptosis regulation, we compared SB-regulated genes to the previously reported PD-target genes.9 Supplementary Tables 1A and B depicted pair-wise comparisons of genes between d3 (differentiation and apoptosis) and d3SB (differentiation with enhanced apoptosis) conditions. Genes, whose expression was similarly modulated by the PD169316 compound, are bolded with an asterisk. Probe sets of interest were retrieved and classified by common biological properties with the NetAffx database annotations.
Remarkable genes involved in the regulation of apoptosis and differentiation are modulated by the SB compound. Increased expression of proapoptotic genes (Clusterin, Bcl2l11, Clic4, Cd24, Ell2) and repression of expression of antiapoptotic genes (Csnk2b, Dyrk1b) were observed. Surprisingly, expression of three potential antiapoptotic genes (Hifα, Ndfip1 and Dusp6) was induced by SB, whereas expression of proapoptotic genes such as Bcl2l11 and Clusterin was increased by the PD compound. This suggests that a subtle balance of pro- and antiapoptotic genes regulates the final status of differentiated cells and/or that the SB compound directly decreases the activity of components participating in a pro-survival pathway, a property not shared by the PD compound.
We also noticed that a significant number of SB-modulated genes were implicated in neuronal differentiation. Indeed, expression of Peripherin 1, Neuronatin, Marcks, Id1, Id2, Dirk1a, Chuck, Top2b and Nestin was induced, whereas expression of rest and nodal, two neuronal repressors, was decreased by the SB compound. Interestingly, many of these genes were also modulated by the PD compound (Supplementary Tables 1A and B). The results obtained with the Affymetrix chips were confirmed by real-time quantitative-polymerase chain reaction (RTQ-PCR) on a selection of SB and PD-regulated genes (Figure 2).

Validation of the microarray data on selected PD- and SB-regulated genes. Total RNAs from ES cells grown without LIF for 3 days (d3), in the absence or presence of 10 μM PD169316 (d3PD) or SB203580 (d3SB), were analyzed by RTQ-PCR with the corresponding specific primers. Fold change ((d3PD/d3) or (d3SB/d3) with standard deviations (line bars)) of at least four independent experiments was plotted for each gene as indicated
Altogether, these results pointed to the fact that the SB and PD chemicals, in addition to modulating the expression of genes involved in apoptosis, might also affect common neuronal differentiation markers, a property potentially linked directly with the p38α MAPK activity. Though, our findings gave us the opportunity to define transcriptional targets of p38α that might correspond to common sets of genes regulated by both inhibitors.
Characterization of p38α modulated-genes
To determine whether genes whose expression level was similarly regulated by the two drugs SB and PD were bona fide p38 targets, we characterized the expression level of selected genes in WT and p38α−/− ES cells grown in the presence or absence of LIF for 3–5 days. As shown in Figure 3, significant variation of gene expression was observed between these two cell lines. Indeed, the expression level of Stra8, Id2 and Cd24a (PD- and SB-induced genes), evaluated during a kinetic of LIF withdrawal, was higher in the p38α−/− cell line in comparison with expression detected in the wild-type (WT) cell line, indicating that the expression of these genes was normally repressed by the p38α pathway. At the opposite side, Lefty1 and Nodal (PD- and SB-repressed genes), whose expression increased upon LIF withdrawal in the WT cell line, was almost undetectable in the p38α−/− cell line, indicating that these genes are induced by the p38 pathway in the WT situation. In contrast, Lef1 (a PD-only repressed gene) is not a transcriptional p38α target as its expression profile is similar in both cell lines. In this study, we have also investigated the expression level of the Bcl-2 gene, previously characterized as a PD-induced gene essential for blocking apoptosis of early precursor cells.8 As shown in Figure 3, an increase of Bcl-2 expression (RNA (Figure 3a) and protein (Figure 3b)) was observed in the p38α−/− cell line deprived of LIF, in good correlation with the increase in BCL2 protein expression in the presence of both p38 inhibitors (Figure 3c). These results demonstrate that Bcl2 is a transcriptional p38α target and that, in differentiated cells, the high level of Bcl-2 expression correlates with low p38 activity, in good agreement with our previous findings.8

Expression profiles of a selection of p38 inhibitor-regulated genes in the WT and p38α−/− ES cell lines. (a) Semiquantitative RT-PCRs were performed with total RNAs from the ES p38α−/− cell line and its parental WT counterpart with the indicated primers. Cells were grown in the presence (+) or absence of LIF for 3, 4 and 5 days (d) as indicated. Western blot analysis with the indicated antibodies with protein cell lysates from (b) the WT and p38α−/− ES cells grown with LIF or without LIF for 3–5 days or from (c) the WT ES cells grown with LIF (+) or without LIF for 4 days in the absence (−4d) or presence of SB (−4dSB) or PD (−4dPD)
The expression of p38α-regulated targets is correlated with the onset of RA-induced neuronal differentiation
To determine whether expression levels of these p38α targets were correlated with the establishment of ES-derived neurons, we have analyzed their expression profiles in the E14TG2a-derived sox1-targeted GFP ES cells, known to differentiate efficiently in functional neurons expressing β3-TUBULIN/TUJ1 and MAP2 (microtubule-associated protein 2) proteins under RA treatment (Li et al.,10 Ying et al.,30 Chung et al.31 and O Feraud, unpublished results).
Flow cytometry experiments were performed at different time points of the kinetic, in the presence of RA (from day 4 to 6 during the EBs formation), with different antibodies directed against NESTIN, a pro-neuronal marker, or TUJ1, which labels neuronal extensions and two MAP2 isoforms, which label dendritic extensions of all types of neurons (MAP2 abc) or of more mature neurons (MAP2 ab) (Figure 4a). We noticed that TUJ1 proteins were present in non-differentiated cells, as previously reported.32 There is a sharp decrease in TUJ1 expression at the first stages of EBs differentiation and a re-expression while cells acquire neuronal fate along with expression of MAP2 isoforms. As shown in Figure 4b, the expression of Bcl2α and β, corresponding to the short and long forms of Bcl2, Cd24a, Id1 and Id2, were upregulated at the same time that neuronal markers (Map2 and Tuj1). In contrast, expression of Nodal, a p38α-dependent neuronal repressor (Pfendler et al.33 and this study), was downregulated while cells acquired neuronal fate. These data suggest that concomitant up- and down-regulated expression of the newly identified p38α-dependent targets could be involved in the establishment of neuronal lineage.

Correlation of the expression profiles of p38α targets and onset of RA-dependent neuronal differentiation. Mouse ES cells (E14TG2a-sox1 targeted EGFP cells) have been induced to differentiate towards the neuronal lineage with RA treatment, leading to enrichment in cells expressing the pro-neuronal (NESTIN) or neuronal markers (β3-TUBULIN/TUJ1 and MAP2). Expression of (a) neuronal markers (quantified by flow cytometry at the time point indicated) and of (b) p38α targets, as indicated (quantified by RTQ-PCR), have been performed at different time points of the kinetics. Means of three independent experiments have been plotted with standard deviations for RTQ-PCR. A representative flow cytometry experiment (over three independent experiments) is shown
Proneuronal effect of BCL2, a p38α transcriptional target
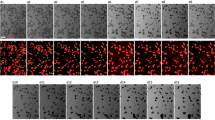
We have previously shown that overexpression of functional antiapoptotic enhanced green fluorescent protein (EGFP)–BCL2 protein does not affect expression of early mesodermal (Brachyury) or pro-neuronal (Nestin) markers during heterogeneous differentiation triggered upon LIF withdrawal for a week.9 However, a slight increase in TUJ1-positive cells was observed by immunolabelling staining (data not shown). To further explore whether this pro-neuronal differentiation effect of BCL2 was additive with repression of p38 activity, we investigated their combined effects on neuronal differentiation. EBs differentiation procedure, as depicted in Figure 5a, was conducted with or without serum, combined with PD or SB treatment, in EGFP (control) or EGFP–BCL2 overexpressor clones. As shown in Figure 5b, the level of expression of two neuronal markers (Tuj1 and Map2) was much higher in the majority of EGFP–BCL2 overexpressor clones treated with PD or SB in comparison to the EGFP control clones, in which a moderate increase in the expression of these markers was observed. In addition, flow cytometry and western blot analyses confirmed a significant increase of TUJ1-expressing cells in the presence of both inhibitors in the EGFP–BCL2 clones compared to the control EGFP clones (Figure 5c, c1, d and f). The majority of EGFP–BCL2 clones do respond to the PD or SB treatments; however, we observed a variation in the expression of markers in the representative selection of clones, as shown in Figure 5c, e and f, which has not yet been further explored. Furthermore, although repression of p38α/β was additive with overexpression of EGFP–BCL2 for TUJ1 expression, we did not observe such an effect on the expression of NESTIN, whose expression level increases by the sole presence of EGFP–BCL2 (Figure 5e and data not shown). Altogether, these results indicate that PD- or SB-dependent repression of p38α/β along with overexpression of EGFP–BCL2, favors neuronal differentiation. In addition, by phase contrast picture analysis, we noticed that the SB-treated cells were more apoptotic than PD-treated cells even in the presence of the EGFP–BCL2 protein. In addition, much less connections between neurons were observed with this compound, and neuronal networks were almost absent in the presence of SB, while these networks were efficiently formed in the presence of PD for the EGFP–BCL2 clones only (data not shown and Figure 5g). Indeed, as shown by immunolabelling experiments, overexpression of EGFP–BCL2 with the PD treatment leads to an increase in TUJ1-positive cells, which form dense organized neuronal networks. These overall results emphasize the importance of repression of p38α to trigger neuronal differentiation and show that overexpression of BCL2, normally induced by p38 repression, is a key actor of this process. Also, the comparison of the effect of the two p38 inhibitors shows that global pro-survival environment (provided by PD) is necessary, along with BCL2 overexpression, for potential neuronal maturation.



Proneuronal effect of EGFP–BCL2 overexpression observed under PD or SB treatment in an EBs-dependent differentiation procedure. (a) Description of the long-term EBs differentiation procedure: EBs formed for 3 days on bacterial Petri dishes in medium without LIF were gently dissociated, plated and cultured on coverslips or culture Petri dishes up to 15 days. When indicated, 10 μM PD169316 or SB203580 was added to the culture media from day 0 to 5 and the serum was removed at day 10. RNA, protein preparation or Immunolabelling was performed at day 15. (b) Quantification, by RTQ-PCR, of Tuj1 and Map2 gene expression in independent EGFP or EGFP–BCL2 clones at day 15. (c) Quantification by flow cytometry of double-positive EGFP/PE cells expressing TUJ1, from independent EGFP and EGFP–BCL2 clones with the PD treatment. (d) Mean values with standard deviation of flow cytometry experiments from (c). Data were analyzed by a Shapiro–Wilk test to determine their belonging to a normal law. Then, they have been treated with an ANOVA test followed by the Tukey test to determine the P-values: lanes 1 versus 2< 0.01, lanes 3 versus 4< 0.05, lanes 1 versus 3: not significant and lanes 2 versus 4< 0.05. (c1) Western blot analysis, on EGFP clone without LIF or serum (lanes 1–3), with PD (lane 2) or with SB (lane 3) and on two independent EGFP–BCL2 clones without LIF or serum (lanes 4–9) with PD (lanes 5 and 8) or with SB (lanes 6 and 9), were performed with the indicated antibodies. ERK2 was used as a constant loading control. (e) Quantification by flow cytometry of double-positive EGFP/PE cells expressing NESTIN, from independent EGFP and EGFP–BCL2 clones with the PD treatment. (f) Quantification by flow cytometry of double-positive EGFP/PE cells expressing TUJ1, from independent EGFP and EGFP–BCL2 clones with the SB treatment. Experiments have been performed twice with at least four independent clones per experiment. Representative clones are shown and a significant increase in the death of EGFP clones with the SB treatment was observed. Only three clones survived the treatment during each of the experiments, which are presented in (f). (g) Immunolabelling of control (EGFP-expressing cells) and EGFP–BCL2-expressing cells at day 15 under the indicated conditions, with an isotypic or the anti-TUJ1 (enlargement: × 100) antibodies. Merged pictures showing EGFP (green), Dapi staining (blue) and isotypic or TUJ1 proteins (red) are presented
Discussion
To study the links between apoptosis and differentiation, we have taken advantage of the availability of p38 MAPK inhibitors, which can prevent (PD 169316) or induce (SB203580) apoptosis in mouse ES-derived differentiated cells. These p38 inhibitors block p38α/β kinase activity at d3, in good agreement with previous reports.34, 35 However, gene expression profiles modulated by these chemicals are far from being similar. Many studies performed with these compounds may now be re-examined in the light of the present data, which demonstrate that these compounds, even if blocking p38α and β isoform activities, may also alter p38-unrelated pathways.
Genes, whose expression is specifically modulated by the SB203580 compound, are involved in many cell differentiation processes (germ cell specification, blood vessel development and various aspects of morphogenesis), cell cycle, cell growth, metabolic pathways, cytoskeleton rearrangements, intracellular transport, protein ubiquitination and chromatin remodelling. We found a puzzling SB-dependent regulation of pro- and antiapoptotic genes that indicates that a subtle balance of expression of these genes leads to global apoptosis. However, our study also revealed a specific SB-dependent downregulation of pluripotent ES cell markers such as Rex1, Fbox15 and Pdgfα.36 Although SB could accelerate cell differentiation and boost apoptosis, by abruptly repressing pluripotent cell markers. At the opposite side, the antiapoptotic PD compound increases expression of survival genes, such as Spp1 gene, recently characterized as a critical self-renewal gene expressed in undifferentiated ES cells.37 In addition, it is noteworthy that none of the genes encoding Ca2+ binding and heavy metal-detoxifying proteins were induced by SB, in contrast to the PD compound, which increases expression of many of such genes. For example, the antiapoptotic Met1 gene, specifically induced by the PD169316 compound and shown to decrease apoptosis of ES-derived differentiated cells,9 was not upregulated by SB203580. This different subset of PD- and SB-modulated genes is most likely related to the differential apoptotic response triggered by these inhibitors.
We have also noticed that a subgroup of the SB-modulated genes is associated with neuronal differentiation. Upregulated genes (such as Nestin, Peripherin1, Neuronatin, Marks and Top2B) are rather pro-neuronal, whereas downregulated genes such as Nodal and Rest block neuronal differentiation.33, 38, 39 Many of these genes are similarly modulated by the PD compound, along with genes involved in the antero-posterior or left–right axis determination of embryos (Nodal, Lefty1 and Tdgf1).40 A subset of these genes, similarly regulated by both p38 inhibitors, are bona fide p38α targets, as revealed by using a p38α−/− ES cell line. We have also shown that the expression of these genes is correlated with the establishment of mature functional neurons induced upon RA treatment. Identification of these direct p38α targets will help to better characterize how waves of p38α activity regulate cell fate decisions. Also, it is of interest to quote that genes such as Id2 (the helix–loop–helix transcription factors of the ids (inhibitor of cell differentiation) gene family) and Bcl2 are under the tight control of p38α only upon LIF withdrawal. Indeed, in pluripotent cells, in which p38α activity is low, these genes are barely expressed. In addition, when overexpressed in pluripotent cells under serum-free conditions, these proteins synergize with LIF and maintain cell pluripotency.41, 42 Altogether, these data shed light on the pleiotropic effects of genes whose functions rely exclusively on cell context. In addition, our results emphasize the key potential role of p38α to control Ids gene expression at specific stages of neuronal maturation.43
Our study shows that the Bcl2 gene, which keeps alive ES-derived differentiated cells, allows preferential differentiation towards the neuronal lineage. Bcl2 belongs to a complex gene family that includes related anti and proapoptotic members. Overexpression of BCL2 protects many cell types against apoptosis triggered by growth factor withdrawal or various oxidative and stress treatments.44 Previous works, performed in neural-crest-derived tumor cell line45 or in the Rat pheochromocytoma PC12 cells, led to the conclusion that BCL2 is involved in neuronal cell differentiation.45, 46 Also, it has been demonstrated that BCL2 contributes to the survival of glial cells or motor neurons.47 Our work shows that bcl2 is a direct target of p38α and that combining overexpression of BCL2 with repression of p38α/β, in serum-free conditions, leads to an increase in Tuj1 and Map2 neuronal markers. In addition, we show that the formation of organized TUJ1+ neuronal cell networks requires the global survival effect provided by the PD compound. It remains to be determined whether genes specifically regulated by PD synergize with BCL2 to form these networks. Together with previous reports demonstrating the importance of the Notch pathway for neuronal commitment,12, 48 this study helps to characterize critical genes that might contribute to the setting up of neuronal lineages. In addition, Bcl2 gene, known as an RA-induced gene,49, 50 could be one of the direct effectors of neuronal differentiation in RA-based differentiation procedures classically used in the ES cell system.
In conclusion, our work presents new data on the characterization of p38α/β inhibitor targets and helps understand their different outcomes on ES-derived differentiated cells. In addition, the identification of p38α targets and in particular of Bcl2 and Nodal, shown respectively to boost (this work) or to repress neuronal lineage,33 give new insights to study the role of p38α during the first steps of differentiation induced upon LIF withdrawal.
Materials and Methods
Cell culture and reagents
S1, CGR8 and E14TG2a (feeder free) and D4 (a D3 subclone, feeder-dependent) ES cell lines (from 129SV mouse) were derived as described5 and grown in DMEM, high glucose (Invitrogen Corporation), supplemented with 0.1 mM β2Mercaptoethanol, 10% FCS, 400 ng/ml gentamycin and LIF (500 pM). The p38α−/− ES cell line, in which both alleles of the p38α gene have been targeted by homologous recombination, has been described previously.27 The Sox1TV2 ES cell line (obtained from Dr. Austin Smith30) is an E14TG2a line in which the Sox1 locus was targeted by homologous recombination to express a GFP–neomycin resistance fusion protein.
The anti-caspase3 (Cell Signaling Technology), anti-extracellular signal-regulated kinase 2 (ERK2) (Santa Cruz), anti-NESTIN (Chemicon), anti-TUJ1 (Covance), anti-MAP2abc (recognizing all MAP2 isoforms; HM-2, Sigma), anti-MAP2ab (adult isoforms of MAP2; AP20, Chemicon), the phycoerythrin (PE) coupled F(ab′)2 fragment Goat anti-Mouse IgG (H+L) (Beckman Coulter), Alexa fluor 594 goat anti-mouse IgG (Molecular probes) and isotypic mouse IgG2A (Sigma) antibodies were used as recommended by the manufacturer. RA (from Sigma) was diluted in EtOH as a 16 mM stock and kept at −20°C.
The SB203580 and PD169316 compounds, diluted in DMSO, were from Calbiochem. Dapi was from Sigma. Fluorescent mounting medium was from Dako-Cytomation.
For microarray experiments, plated ES cells (after two passages without feeders for D4 cell line) were diluted at 105 cells/ml in ES cell medium with or without LIF in the presence or absence of 10 μM SB203580. Cell medium was changed every day and mRNAs were prepared on the third day (d3).
Differentiation procedure and apoptotic test
ES cells were plated (105 cells/ml) in LIF-containing medium. On the next day, the plated cells were washed with PBS and fed for the indicated times (3–5 days) with medium supplemented or not with LIF. When indicated, 10 μM PD169316 or 10 μM SB203580 were added from day 0 to 4, without LIF. The medium was changed every day. Apoptosis was detected by western blot analysis with anti-caspase 3 antibody, which reveals the full length or cleaved activated form of this caspase.
For long-term differentiation, ES CGR8 control cells (expressing EGFP) and EGFP–BCL2 clones were grown as EBs (1.5 × 105 cells/ml) on bacterial Petri dishes in medium without LIF during 3 days. EBs were then gently dissociated, plated and cultured on gelatinized coverslips or cell culture dishes up to 15 days. When indicated, 10 μM PD169316 or 10 μM SB203580 were added to the culture media from day 0 to 5 and the serum was removed from day 10 to 15. Medium was changed every day from day 0 to 5 and every other day up to day 15. Immunolabelling staining, flow cytometry, proteins or RNA preparations were performed at day 15.
Neuronal differentiation was also induced with RA treatment, as previously described, via EBs differentiation.30 Briefly, Sox1TV2 ES cells were allowed to form EBs in liquid culture condition onto non-adherent dishes during 8 days with a 48 h RA stimulation (10−6 M) between days 4 and 6. At day 8 of differentiation, EBs were dissociated and the cells plated on poly-D-lysine/laminin-coated dishes. During the first 48 h of plating, bFGF (10 ng/ml, R&D Systems) has been added to the culture medium, to increase the number of neuronal progenitor cells.
Cell lysates, western blots and immunohistochemistry
Plated cells were rinsed twice with room temperature PBS and lysed directly in mild RIPA buffer (PBS, 1% triton, 1% NP40, 0.05% SDS, 1 μg/ml protease inhibitor cocktail, 1 mM Pefabloc, 20 mM NaF, 1 mM Na vanadate) and centrifuged for 10 min at 10 000 r.p.m. Cell lysates were resolved by SDS-PAGE (10 or 12% (for caspase)) and electro-transferred onto nitrocellulose membranes in the presence or absence (for caspase) of 0.07% SDS. Proteins were reacted with the different antibodies, as recommended by the manufacturers.
Immunolabelling of cells with anti-TUJ1 or isotypic antibodies were carried out as follows: cells were grown on coverslips under the indicated conditions. Cells were fixed with 4% formaldehyde (diluted in PBS) for 10 min at room temperature and permeabilized with 0.1% Triton X-100 for 20 min. Fixed cells were rinsed with PBS and incubated with blocking buffer (PBS, 3% bovine serum albumin (BSA), 0.1% Triton X-100) for 1 h, followed by incubation with the primary antibody (anti-TUJ1 diluted 1/300, isotypic antibody diluted 1/100) at room temperature for 1 h in PBS with 0.5% BSA and 0.1% Triton X-100. After washing with PBS, the cells were incubated for 1 h with species-specific fluorescence-labelled secondary antibodies diluted to 1/6000. Samples were washed twice with PBS, counterstained with Dapi at 100 ng/ml for 2 min and observed under a mounting medium. Immunofluorescent pictures (magnifications × 100) were recorded by computer acquisition using the Quips smart capture Visys software with the Olympus AX70 microscope.
Flow cytometry experiments
Plated cells were rinsed once with room temperature PBS and treated for 15 min at 37 °C with a 50 mM EDTA/PBS solution. The cells were flushed using a micropipette and centrifuged for 2 min at 2000 r.p.m. The pellet was then fixed with 4% formaldehyde in PBS for 20 min at room temperature. Fixed cells were rinsed with PBS (centrifugation for 5 min at 3000 r.p.m.) and incubated with blocking buffer (PBS, 0.25% saponin, 1% BSA) for 5 min at room temperature followed by incubation at 4°C with the primary antibody diluted 1/200 for 45 min in blocking buffer. Cells were then incubated for 45 min at 4°C with species-specific fluorescence-labelled secondary antibodies diluted to 1/200. Samples were washed once with PBS/0.25% saponin, 0.1% Tween20 and resuspended in PBS. The samples were then analyzed using a FacsCanto device (BD Bioscience).
Semiquantitative RT-PCR and RTQ-PCR experiments
Total RNAs from adherent ES cells were prepared with the Trizol reagent kit7 and treated with DNAse (5 U/50 μg RNA, Sigma). Total RNA (1 μg) was reverse-transcribed with random hexameric primers and the MLV Reverse Transcriptase (Sigma). The RT reaction products were used for PCR reactions with specific primer sets as previously described. RTQ-PCR experiments have been performed with the Light Cycler system (Roche, Idaho Technologies).9 The Cp value (minimum cell cycle number in the linear zone of amplification) was determined for each gene in these conditions: 3 days without LIF (d3) or 3 days without LIF and 10 μM PD169316 (d3PD) or 10 μM SB203580 (d3SB). The FC (fold change), corresponding to the level of induction or repression of gene expression in the presence of PD169316 or SB203580, was calculated with this formula: 2(Cp (d3)-Cp (d3PD)) or : 2(Cp (d3)-Cp (d3SB)).
For quantification of p38α target genes in EBs neuronal differentiation (Figure 4b), RTQ-PCR was similarly performed on a Lightcycler equipment using amplicon probes designed with QuantiTect Primer Assay (Qiagen): Cd24a: ref. QT00122850; Id1: ref. QT00240219; Id2: ref. QT01038870; Nodal: ref. QT00125685; Bcl2va1/Bcl2α: ref QT01057217; Bcl2vb1/Bcl2β: ref QT01057224. Normalization of data was performed with ribosomal protein RL37A gene: ref. QT 00252266.
Quantification of neuronal markers (Figure 5b) by RTQ-PCR was performed with the MX4000 system (Stratagene). Increasing amounts of cDNA were treated with each primer pair in duplicate resulting in a standard curve for each pair of primers. Efficiency of PCR and correlation coefficient has been deduced from this curve allowing the validation of the primers, when efficiency was superior to 85% and inferior to 110% and correlation coefficient (r2) superior to 0.985. The Ct value (minimum cell cycle number in the linear zone of amplification) was determined for each gene in these conditions: 15 days without LIF and serum without (−L−S) or with 10 μM PD169316 (−L−S+PD) or 10 μM SB203580 −L−S+SB). The relative expression of the gene of interest was deduced of the Ct using the ΔΔCt method with Hprt house-keeping gene as the reference.

Expression of the result in percentage of relative expression considering that the efficiency of primers is quite the same and close to 1.
Sequences of primers used for all the genes tested in RT-PCR or RTQ-PCR are in Supplementary Figure 1.
Microarray experiments: hybridization and data analysis
Total RNAs from adherent ES cells were prepared with the Qiagen column kit (Qiagen) and treated with DNAse (5 U/50 μg RNA, Sigma). The complete procedure of gene array experiments have been previously described.9
Microarray data have been submitted to and approved by the GEO public library, NCBI, http://www.ncbi.nlm.nih.gov/geo/submission/login/, under the access number: GSE2042.
Expression vectors and stable ES clones
Derivation of stable clones expressing EGFP and EGFP–BCL2 proteins (from the pCXN2 vector) has been previously described.8, 9 Experiments were performed at least three times on four to six independent homogeneous fluorescent clones (95–98% EGFP+, as determined by flow cytometry, data not shown).
Abbreviations
- EBs:
-
embryoid bodies
- ES:
-
embryonic stem
- ERK2:
-
extracellular-signal regulated kinase 2
- EGFP:
-
enhanced green fluorescent protein
- LIF:
-
leukemia inhibitory factor
- MAP2:
-
microtubule-associated protein 2
- MAPK:
-
mitogen-activated protein kinase
- RA:
-
retinoic acid
- RTQ-PCR:
-
real-time quantitative-polymerase chain reaction
References
Shellard J, Perreau J, Brulet P . Role of leukemia inhibitory factor during mammalian development. Eur Cytokine Netw 1996; 7: 699–712.
Burdon T, Smith A, Savatier P . Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol 2002; 12: 432.
Metcalf D . The unsolved enigmas of leukemia inhibitory factor. Stem Cells 2003; 21: 5–14.
Babinet C, Cohen-Tannoudji M . Genome engineering via homologous recombination in mouse embryonic stem (ES) cells: an amazingly versatile tool for the study of mammalian biology. An Acad Bras Cienc 2001; 73: 365–383.
Smith AG . Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev Biol 2001; 17: 435–462.
Wobus AM, Boheler KR . Embryonic stem cells: prospects for developmental biology and cell therapy. Physiol Rev 2005; 85: 635–678.
Duval D, Reinhardt B, Kedinger C, Boeuf H . Role of suppressors of cytokine signaling (Socs) in leukemia inhibitory factor (LIF) -dependent embryonic stem cell survival. FASEB J 2000; 14: 1577–1584.
Duval D, Malaise M, Reinhardt B, Kedinger C, Boeuf HA . p38 inhibitor allows to dissociate differentiation and apoptotic processes triggered upon LIF withdrawal in mouse embryonic stem cells. Cell Death Differ 2004; 11: 331–341.
Duval D, Trouillas M, Thibault C, Dembele D, Diemunsch F, Reinhardt B et al. Apoptosis and differentiation commitment: novel insights revealed by gene profiling studies in mouse embryonic stem cells. Cell Death Differ 2006; 13: 564–575.
Li M, Pevny L, Lovell-Badge R, Smith A . Generation of purified neural precursors from embryonic stem cells by lineage selection. Curr Biol 1998; 8: 971–974.
Ying QL, Smith AG . Defined conditions for neural commitment and differentiation. Methods Enzymol 2003; 365: 327–341.
Bouhon IA, Kato H, Chandran S, Allen ND . Neural differentiation of mouse embryonic stem cells in chemically defined medium. Brain Res Bull 2005; 68: 62–75.
Gossrau G, Thiele J, Konang R, Schmandt T, Brustle O . Bone morphogenetic protein-mediated modulation of lineage diversification during neural differentiation of embryonic stem cells. Stem Cells 2007; 25: 939–949.
Aouadi M, Bost F, Caron L, Laurent K, Le Marchand Brustel Y, Binetruy B . p38 mitogen-activated protein kinase activity commits embryonic stem cells to either neurogenesis or cardiomyogenesis. Stem Cells 2006; 24: 1399–1406.
Keren A, Tamir Y, Bengal E . The p38 MAPK signaling pathway: a major regulator of skeletal muscle development. Mol Cell Endocrinol 2006; 252: 224–230.
Binetruy B, Heasley L, Bost F, Caron L, Aouadi M . Concise review: regulation of embryonic stem cell lineage commitment by mitogen-activated protein kinases. Stem Cells 2007; 25: 1090–1095.
Kumar S, Boehm J, Lee JC . p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2003; 2: 717–726.
Abraham RT . MAPKAP kinase-2; three's company at the G(2) checkpoint. Mol Cell 2005; 17: 163–164.
Horstmann S, Kahle PJ, Borasio GD . Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J Neurosci Res 1998; 52: 483–490.
Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X et al. p38 kinase is activated in the Alzheimer's disease brain. J Neurochem 1999; 72: 2053–2058.
Culbert AA, Skaper SD, Howlett DR, Evans NA, Facci L, Soden PE et al. MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity. Relevance to neuroinflammation in a transgenic mouse model of Alzheimer disease. J Biol Chem 2006; 281: 23658–23667.
Niu W, Huang C, Nawaz Z, Levy M, Somwar R, Li D et al. Maturation of the regulation of GLUT4 activity by p38 MAPK during L6 cell myogenesis. J Biol Chem 2003; 278: 17953–17962.
Pfeiffer ZA, Aga M, Prabhu U, Watters JJ, Hall DJ, Bertics PJ . The nucleotide receptor P2X7 mediates actin reorganization and membrane blebbing in RAW 264.7 macrophages via p38 MAP kinase and Rho. J Leukoc Biol 2004; 75: 1173–1182.
Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R . Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol 2004; 24: 6539–6549.
Lee ER, McCool KW, Murdoch FE, Fritsch MK . Dynamic changes in histone H3 phosphoacetylation during early embryonic stem cell differentiation are directly mediated by mitogen- and stress-activated protein kinase 1 via activation of MAPK pathways. J Biol Chem 2006; 281: 21162–21172.
Saklatvala J . The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol 2004; 4: 372–377.
Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel CA . Deficiency of the stress kinase p38alpha results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J Exp Med 2000; 191: 859–870.
Ihle JN . The challenges of translating knockout phenotypes into gene function. Cell 2000; 102: 131–134.
Aouadi M, Binetruy B, Caron L, Le Marchand-Brustel Y, Bost F . Role of MAPKs in development and differentiation: lessons from knockout mice. Biochimie 2006; 88: 1091–1098.
Ying QL, Stavridis M, Griffiths D, Li M, Smith A . Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol 2003; 21: 183–186.
Chung S, Shin BS, Hedlund E, Pruszak J, Ferree A, Kang UJ et al. Genetic selection of sox1GFP-expressing neural precursors removes residual tumorigenic pluripotent stem cells and attenuates tumor formation after transplantation. J Neurochem 2006; 97: 1467–1480.
Ginis I, Luo Y, Miura T, Thies S, Brandenberger R, Gerecht-Nir S et al. Differences between human and mouse embryonic stem cells. Dev Biol 2004; 269: 360–380.
Pfendler KC, Catuar CS, Meneses JJ, Pedersen RA . Overexpression of Nodal promotes differentiation of mouse embryonic stem cells into mesoderm and endoderm at the expense of neuroectoderm formation. Stem Cells Dev 2005; 14: 162–172.
Davies SP, Reddy H, Caivano M, Cohen P . Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 2000; 351: 95–105.
Avitzour M, Diskin R, Raboy B, Askari N, Engelberg D, Livnah O . Intrinsically active variants of all human p38 isoforms. FEBS J 2007; 274: 963–975.
Palmqvist L, Glover CH, Hsu L, Lu M, Bossen B, Piret JM et al. Correlation of murine embryonic stem cell gene expression profiles with functional measures of pluripotency. Stem Cells 2005; 23: 663–680.
Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C et al. Dissecting self-renewal in stem cells with RNA interference. Nature 2006; 442: 533–538.
Jones FS, Meech R . Knockout of REST/NRSF shows that the protein is a potent repressor of neuronally expressed genes in non-neural tissues. Bioessays 1999; 21: 372–376.
Greenway DJ, Street M, Jeffries A, Buckley NJ . RE1 silencing transcription factor maintains a repressive chromatin environment in embryonic hippocampal neural stem cells. Stem Cells 2007; 25: 354–363.
Lu CC, Robertson EJ . Multiple roles for Nodal in the epiblast of the mouse embryo in the establishment of anterior-posterior patterning. Dev Biol 2004; 273: 149–159.
Ying QL, Nichols J, Chambers I, Smith A . BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003; 115: 281–292.
Yamane T, Dylla SJ, Muijtjens M, Weissman IL . Enforced Bcl-2 expression overrides serum and feeder cell requirements for mouse embryonic stem cell self-renewal. Proc Natl Acad Sci USA 2005; 102: 3312–3317.
Peddada S, Yasui DH, LaSalle JM . Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum Mol Genet 2006; 15: 2003–2014.
Cory S, Adams JM . The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647–656.
Zhang KZ, Westberg JA, Holtta E, Andersson LC . BCL2 regulates neural differentiation. Proc Natl Acad Sci USA 1996; 93: 4504–4508.
Suzuki A, Tsutomi Y . Bcl-2 accelerates the neuronal differentiation: new evidence approaching to the biofunction of bcl-2 in the neuronal system. Brain Res 1998; 801: 59–66.
Sato N, Sakuma C, Kato H, Milligan CE, Oppenheim RW, Yaginuma H . Bcl-2 rescues motoneurons from early cell death in the cervical spinal cord of the chicken embryo. J Neurobiol 2002; 53: 381–390.
Lowell S, Benchoua A, Heavey B, Smith AG . Notch promotes neural lineage entry by pluripotent embryonic stem cells. PLoS Biol 2006; 4: e121.
Sumantran VN, Brederlau A, Funa K . BMP-6 and retinoic acid synergistically differentiate the IMR-32 human neuroblastoma cells. Anticancer Res 2003; 23: 1297–1303.
Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, Isogai E et al. Bcl-2 is a key regulator for the retinoic acid-induced apoptotic cell death in neuroblastoma. Oncogene 2006; 25: 5046–5055.
Acknowledgements
We thank all members of the UMR-CNRS-5164-CIRID and, in particular, Jean François Moreau for helpful insights and a careful reading of the paper. We thank C Gabel for the gift of the ES WT and ES p38α−/− cell lines, A Smith for the gift of the ES CGR8 WT and E14TG2aSox1-EGFP ES cell lines, A Dierich for the gift of the S1 ES cell line, R Kemler for the gift of the D3 ES cell line and Dr Miyazaki for the pCXN2 expression vector. We thank V Pitard, M Landry and B Guillotin, respectively, for very good advices concerning the flow cytometry experiments, the characterization of neuronal cells and RTQ-PCR experiments. Thanks also to E Préchais for preliminary data concerning the validation of the p38α target genes. HB thanks P Dubus for constant support and reading of the paper. This work was founded by the European consortium FungenES (6th Framework, project no LSHG-CT-2003-503494, Coordinator: Professor J Hescheler), CNRS, the Ministere of Research (Affymetrix microarray grant, IGBMC genopole platform, Strasbourg), University of Bordeaux 2, the Ligue National contre le Cancer, Comités Aquitaine-Charentes, the Region Aquitaine and the IFR66. MT, OF and MG were financed by a FungenES fellowship and CS by a Region Aquitaine fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by R De Maria
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Trouillas, M., Saucourt, C., Duval, D. et al. Bcl2, a transcriptional target of p38α, is critical for neuronal commitment of mouse embryonic stem cells. Cell Death Differ 15, 1450–1459 (2008). https://doi.org/10.1038/cdd.2008.63
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.63
Keywords
This article is cited by
-
Phospholipase D1 Signaling: Essential Roles in Neural Stem Cell Differentiation
Journal of Molecular Neuroscience (2018)
-
MicroRNA-128-3p Protects Mouse Against Cerebral Ischemia Through Reducing p38α Mitogen-Activated Protein Kinase Activity
Journal of Molecular Neuroscience (2017)
-
Scutellarin Alleviates Behavioral Deficits in a Mouse Model of Multiple Sclerosis, Possibly Through Protecting Neural Stem Cells
Journal of Molecular Neuroscience (2016)
-
Phospholipase D1 Increases Bcl-2 Expression During Neuronal Differentiation of Rat Neural Stem Cells
Molecular Neurobiology (2015)
-
A p38mapk-p53 cascade regulates mesodermal differentiation and neurogenesis of embryonic stem cells
Cell Death & Disease (2013)
