
Abstract
Acute myeloid leukaemia (AML) is a molecularly and clinically heterogeneous disease, and its incidence is increasing as the populations in Western countries age. Despite major advances in understanding the genetic landscape of AML and its impact on the biology of the disease, standard therapy has not changed significantly in the last three decades. Allogeneic haematopoietic stem cell transplantation remains the best chance for cure, but can only be offered to a minority of younger fit patients. Molecularly targeted drugs aiming at restoring apoptosis in leukaemic cells have shown encouraging activity in early clinical trials and some of these drugs are currently being evaluated in randomised controlled trials. In this review, we discuss the current development of drugs designed to trigger cell death in AML.
Similar content being viewed by others

Main
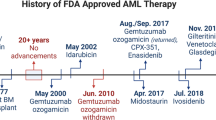
Acute myeloid leukaemia (AML) is characterised by the rapid proliferation of immature myeloid progenitors resulting in the suppression of normal haematopoiesis. Standard therapy includes intensive chemotherapy, followed in patients with poor prognostic features by high dose chemotherapy followed by allogeneic bone marrow transplant (Ferrara and Schiffer, 2013). However, because of the associated morbidity, intensive chemotherapy followed by allogeneic transplant is only feasible in young patients. From an epidemiological perspective however, AML affects primarily patients older than 60 years, who are not eligible for such treatments whereas their prognostic is overall poor without long-term survival (Ossenkoppele and Löwenberg, 2015). There is therefore an unmet medical need to develop new therapies for AML patients.
From the molecular point of view, AML is thought to develop through a multistep process progressing through the acquisition by tumour cells of multiple genomic alterations, which affect several cellular parameters. Genomic alterations in AML have classically been classified in those inducing a block in differentiation, those inducing proliferation and more recently those affecting epigenetic control (Döhner et al, 2010; Fathi and Abdel-Wahab, 2012). This classification has recently been challenged by data suggesting that alterations do not happen independently of each other but rather cooperate in disease progression (Shih et al, 2015).
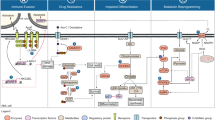
Apoptosis is one of the mechanisms of regulated cell death and has been the focus of intensive research over the last century. Evasion from apoptosis is a required step for malignant tumour progression (Hanahan and Weinberg, 2011). Apoptosis is accomplished through two separate but connected pathways: the intrinsic pathway which converges on the mitochondria and leads to the formation of the apoptosome and caspase-9 activation; and the extrinsic pathway which transduces signalling from external apoptotic cues (see Figure 1).

Schematic representation of the intrinsic and extrinsic pathways of apoptosis.
Activation of the intrinsic pathway is under the control of the BCL2 family of protein, which can be divided into anti-apoptotic proteins (BCL2, BCL-XL, BCL-W (BCL2L2), myeloid cell leukaemia sequence 1 (MCL1) et BFL1/A1 (BCL2A1)), pro-apoptotic BH3 only proteins (BH3-interacting domain death agonist (BID), BCL-2 antagonist of cell death (BAD), BCL-2-interacting killer (BIK), PUMA (BCL-2-binding component 3 (BBC3)), NOXA (phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1)), BCL2-modifying factor (BMF) and HRK) and pro-apoptotic effector proteins (BCL-2-associated X protein (BAX), BCL-2-antagonist/killer (BAK) and BCL-2-related ovarian killer (BOK)). Upon apoptotic stimuli, BH3-only proteins are upregulated (e.g. PUMA and NOXA upon p53 activation), and anti-apoptotic BCL2 family proteins are downregulated, leading to change in the balance of pro- vs anti-apoptotic BCL2 family proteins. Other BH-3 only proteins can be induced by various stimuli, such as BIM which is stabilised in response to E2F1. This unbalance leads to activation of the effector proteins BAK and BAX, which assemble into multimeric pores in the mitochondrial membrane, lead to mitochondrial outer membrane permeabilisation and cytochrome c release into the cytosol (Ashkenazi et al, 2017). The release of cytochrome c leads to the formation of the apoptosome via recruitment of APAF1 and pro-caspase-9, followed by activation of caspase-9 by proteolytic cleavage. Caspase-9 activates caspase-3 and -7 by proteolysis.
The extrinsic pathway comprises the death receptors DR4 and DR5 as well as FAS, which are members of the TNF-receptor family, and share the common trait of having a death domain. This allows the formation of a death-inducing signalling complex by recruiting FAS-associated Death Domain and pro-caspase-8, which leads to the proteolytic activation of caspase-8, which in turn activates caspase-3 and -7. The extrinsic pathway is regulated by several inhibitor of apoptosis proteins (IAPs) which inhibit the transmission of the apoptotic signal at different levels. Eight IAP family members have been identified in humans: XIAP (X-linked inhibitor of apoptosis), cellular inhibitor of apoptosis 1 (cIAP-1), cIAP-2, survivin, NAIP (neuronal apoptosis inhibitory protein), livin, BRUCE/Apollon and IAP-like protein 2. Their grouping as a family is linked to the presence of 1–3 baculovirus IAP repeats (BIR) domain. XIAP cIAP1 (BIRC2), cIAP2 (BIRC3) are the sole members of the family functioning as inhibitors of apoptosis and XIAP is the only direct inhibitor of caspases 3, 7 and 9. Cellular IAP (cIAP) 1 and 2 inhibit caspases by targeting them for ubiquitin-mediated proteasomal degradation which as consequences in apoptosis but also other signalling events: for example, modulation of caspase 8 stability influences the transmission of apoptotic signals by the death-inducing signalling complex, but also modulates the NF-κB survival pathway. Several naturally occurring inhibitors of IAP have been identified, such as Smac/DIABLO (second mitochondrial-derived activator of caspases/direct IAP binding protein with low PI), Omi/HrtA2 (HTRA serine peptidase 2) and XAF-1 (XIAP-associated factor 1), but among these, Smac/DIABLO appears to be the strongest IAP inhibitor (Obexer and Ausserlechner, 2014).
Aside from the apoptotic machinery, another protein that plays a central role in regulating apoptosis is the p53 protein, which integrates a number of signals resulting from various cellular insults and induces adequate cellular response by inducing cell-cycle arrest, repair and/or apoptosis. Termed the ‘guardian of the genome’, p53 regulates cell fate following several types of DNA insults. Two different outcomes can be mediated by activated p53: cell cycle arrest (and DNA repair) or apoptosis (if DNA repair is not possible). Cell cycle arrest following p53 activation is mediated by p21 (WAF1/CIP1, coded by the tumour suppressor gene CDKN1A), while PUMA, NOXA and BIM are the main mediators of p53-induced apoptosis (Yu et al, 2001; Villunger et al, 2003; Schlereth et al, 2010).
Identifying strategies to induce or restore defective apoptosis is therefore a high priority challenge for cancer therapy (Fesik, 2005). The apoptotic pathways can be therapeutically targeted at several levels, which can be grouped into two broad mechanisms: (i) induction of apoptosis, for example by restoring active p53, or by activating death receptors or (ii) restoration of downstream signalling cascades. Unlike many solid tumours, in which one of the most common events leading to a block in apoptosis is a genomic loss of p53 by either deletion or mutation, the TP53 locus is wild-type in most cases of non-complex karyotype de novo AML (Haferlach et al, 2008; Rücker et al, 2012; Cancer Genome Atlas Research Network, 2013), suggesting that alternate mechanisms are at play to block apoptosis in the majority of AML cases.
In this review, we discuss alterations in apoptosis cascades underlying AML and the current development of drugs designed to trigger cell death in these malignancies.
Mechanisms of dysregulation of apoptosis in AML
Apoptosis is dysregulated in virtually all malignancies but the means of dysregulation and the signalling elements involved are highly variable from one tumour type to another. For example, although the tumour suppressor TP53 is the most commonly mutated gene in human solid tumours, genomic inactivation of TP53 is much less common in haematological malignancies (Hainaut and Pfeifer, 2016). When interrogating the cbioportal.org repository (www.cBioportal.org– Cerami et al, 2012; Gao et al, 2013), TP53 gene alterations are found in 2.8–10.6% of adult leukaemia and in about 9% of AML cases (Cancer Genome Atlas Research Network, 2013). Interestingly, TP53 alterations in AML are associated with distinct genomic and biological characteristics, such as complex karyotype and increased genomic instability, which correlate with poor prognosis (Haferlah et al, 2008; Rücker et al, 2012). This observation has led to the identification of ‘AML with TP53 mutations, chromosomal aneuploidy, or both’ as a separate prognostic subgroup, encompassing the previously identified subgroup of patients with complex karyotype AML (Papaemanuil et al, 2016). While most commonly associated with resistance to chemotherapy, TP53 alterations were recently shown to be associated with improved response rate in patients treated with decitabine (Welch et al, 2016). Interestingly, in this study, survival was similar in patient with poor risk cytogenetic features/TP53 mutation and patients with intermediate-risk cytogenetics/wild-type TP53.
The frequency of TP53 gene alterations remains low in de novo and/or non-complex karyotype AML. However, functional inactivation of p53 or of its pathway appears to be a requisite for transformation; loss of p53 function in cancer cells with wild-type TP53 is often caused by abnormalities in p53-regulatory proteins, including overexpression of mouse double minute 2 (MDM2)/MDMX, deletion of CDKN2A/ARF, and alterations in ATM. Contrary to solid tumours, the CDKN2A locus and ATM are rarely altered in AML. Likewise, MDM2 amplification is rare in AML, but its overexpression has been shown in several studies and correlates with shorter progression-free survival (Faderl et al, 2000). MDM2 overexpression also correlates with wild-type TP53 gene status and loss of p21WAF1/CIP1 expression (Quintás-Cardama et al, 2017) which supports its pathological implication as a mean to escape apoptosis.
Downstream effectors of the intrinsic apoptotic pathway are also deregulated in AML, but similar to the case of MDM2, genomic alterations are rare. This may be explained by a founding role of genomic alteration of epigenetic regulators in AML (Papemmanuil et al, 2016) which may lead to deregulated expression without genetic alteration. Thus, BCL2 and other members of the BCL2 family of proteins have been reported to be overexpressed in AML and correlate with resistance to cytotoxic chemotherapy and to targeted agents (Kornblau et al, 1999; Mehta et al, 2013). More recently, overexpression of BCL-XL (BCL2L1) and MCL1 were shown to also play a crucial role in AML pathogenesis (Xiang et al, 2010; Glaser et al, 2012).
The role of the extrinsic apoptotic pathway in AML appears less clear. Here again no recurrent genetic alterations have been reported, but leukaemic blasts were shown to decrease or lose FAS expression during transformation from myelodysplastic syndrome to AML and this correlates with increased methylation of the FAS promoter. Treatment with decitabine was shown to induce FAS expression as well as DR5 expression and was suggested to be one of the mechanisms of action of demethylating agents (Ettou et al, 2013; Karlic et al, 2014). IAPs are also important mediators of resistance to therapy in AML. IAP proteins have been shown to be expressed in samples from AML patients and higher expression correlated with lack of complete response (CR) following induction with standard (intensive) chemotherapy (Pluta et al, 2015).
Targeting the MDM2/p53 pathway
MDM2 regulates p53 stability via ubiquitination, which promotes p53 degradation by the proteasome. MDMX on the contrary has no intrinsic ubiquitin ligase activity and may act on p53 by heterodimerising with MDM2, thereby enhancing its ubiquitin ligase activity. The regulation of MDM2 and MDMX levels is complex but their expressions are increased by oncogenic signals. It should be noted that both of these genes are direct p53 transcriptional targets, though MDM2 is more broadly responsive to p53 activation (Phillips et al, 2010). The discovery of the nutlin family of compounds, which are small molecules able to disrupt the MDM2-p53 interaction, by Vassilev et al in 2004 (Vassilev et al, 2004) opened the way for efficiently targeting this pathway in the clinic. Other classes of agents have since then been shown to be able to disrupt this interaction (Ding et al, 2006) and several MDM2 antagonists are currently in clinical development in solid tumours and haematological malignancies. These compounds induce p53 stabilisation and induction of its target genes (CDKN1A and PUMA among others). Many studies have shown the efficacy of MDM2 inhibitors in preclinical models of AML, either alone or combined with various inhibitors, including those targeting the mitogen-activated protein kinase pathway (Long et al, 2010; Zang et al, 2010; Weisberg et al, 2015; Lehmann et al, 2016). These preclinical studies have shown that approximately 50–60% of AML cell lines and patients-derived AML samples are sensitive to MDM2 inhibition (Long et al, 2010; Weisberg et al, 2015). As expected given the mode of action of MDM2 inhibitors, TP53 mutated cells are intrinsically resistant to this approach.
The available clinical data regarding MDM2 inhibitors in AML is currently limited to phase 1 trial data. The first MDM2 antagonist used in the clinic was RG7112 (Roche, NJ, USA), a nutlin derivative given orally. The recently published phase 1 study of RG7112 in patients with leukaemia showed that this agent had meaningful clinical activity in AML, but not in other types of leukaemia, with 5 of 30 evaluable AML patients having an International Working Group (IWG)-defined response (Andreeff et al, 2016). Interestingly, two patients with TP53-mutant AML displayed transient clinical response. The main adverse events were gastrointestinal toxicity, comprising nausea, vomiting and diarrhoea, which were dose-limiting and bone marrow suppression resulting in neutropenia, febrile neutropenia and thrombocytopenia. The maximum tolerated dose of RG7112 in patients with AML was declared at 1500 mg BID for the first 10 days of 28-day cycles. Pharmacokinetic analysis in this study showed that the mean exposure at MTD was slightly lower than the expected efficacious exposure in animal models. This prompted Roche to develop a second generation, more potent MDM2 inhibitor (RG7388–idasanutlin), which has been tested as an orally given drug in patients with solid tumours and as an oral and intravenous formulation in patients with AML. In these studies, the safety profile of RG7388 was in fact comparable to that of RG7112, suggesting that the observed adverse events result from on-target effects on the gastrointestinal tract and the bone marrow (Siu et al, 2014). The safety profile is consistent across several pharmacological classes of MDM2 inhibitors tested in the clinic (Kurzrock et al, 2012; Wagner et al, 2015), confirming on-target effects. The use of an intravenous formulation limited the impact of gastrointestinal toxicity on dosing and exposure, and interesting results have been reported with idasanutlin intravenously administered with cytarabine (Yee et al, 2014; Reis et al, 2016). The recommended phase 2 dose (RP2D) of RG7388 administered orally was 600 mg BID for both the single agent and the combination arm. Six of 29 patients (21%) treated with single agent RG7388 achieved a CR or CRi/MLFS, while 11/46 patients (24%) treated with RG7388+cytarabine had a CR or CRi. Based on these data, a phase 3 trial of idasanutlin in combination with intermediate dose cytarabine in patients with refractory/relapsed AML is currently ongoing (NCT02545283). Interestingly, in those studies, pre-treatment MDM2 expression using flow cytometry correlated with clinical response suggestive of oncogene addiction. MDM2 expression may thus represent a valuable biomarker for the selection of AML patients who may be candidates for idasanutlin therapy (Reis et al, 2016). Safety and efficacy of another MDM2 inhibitor (MK-8242) in AML patients was reported recently. In this study, 26 patients with AML received MK-8242 orally twice daily. The maximum administered dose was 300 mg BID for 7 days followed by 14 days of rest. Of 24-efficacy evaluable patients, one had a CR. Unfortunately, the study, which was initially designed to assess the safety and efficacy of MK-8242 alone and in combination with cytarabine, was closed prematurely due to pipeline prioritisation (Ravandi et al, 2016). Other MDM2 inhibitors, such as HDM201 (Novartis Pharma, Basel, Switzerland), are currently in phase 1 trials in patients with AML, but no data have yet been reported. One of the potential limitations to the use of MDM2 inhibitors is the emergence of TP53 mutations as a mechanism of resistance (Jung et al, 2016), while TP53 mutant cells may also be selected for by cytotoxic chemotherapy (Wong et al, 2015).
Targeting the BCL2 family in AML
B-cell lymphoma 2 (BCL2) was discovered on B-cell leukaemia and follicular lymphomas more than 30 years ago (Fukuhara and Rowley, 1978; Tsujimoto et al, 1984). Although it was characterised as a bonafide oncogene, the mechanisms by which BCL-2 induces transformation (namely by blocking apoptosis) were only understood later. Within the following decade, a dozen of structurally related proteins were described. As introduced earlier, these proteins can be classified into three different groups: (i) the multidomain anti-apoptotic proteins such as BCL2, BCL-XL and MCL1 for example, (ii) the multidomain pro-apoptotic effector proteins such as BAX or BAK and (iii) the BH3-only group of pro-apoptotic proteins. This lalter group can be further subdivided into activator proteins, such as BIM, BID or PUMA, and sensitisers, such as BAD, BIK or NOXA for example (reviewed in Letai, 2008 and Hata et al, 2015). The major role of both BCL-2 and BCL-XL in promoting survival of cancer cells has made these proteins interesting targets for specific inhibition. MCL1 has also emerged as a mechanism of resistance to apoptosis and to BCL-2/BCL-XL inhibitors, and as such is considered a potential therapeutic target (Kotschy et al, 2016). So far, the number of BCL-2/BCL-XL inhibitors that have entered the clinic is limited and results in AML have been heterogeneous. Many compounds initially depicted as BH3-mimetic or BCL2 inhibitors have failed to show selectivity and actual binding to BCL2-related proteins in cells (van Delft et al, 2006; Soderquist and Eastman, 2016). However, several second-generation Bcl-2-specific inhibitors are currently in later stages of development in B-cell malignancies and venetoclax was recently approved for the treatment of patients with TP53 deleted chronic lymphocytic leukaemia.
Obatoclax, a first-generation pan-Bcl-2 family antagonist (putative BH3 mimetics) was assessed in phase I/II study in patients older than 70 years with treatment naive AML (Schimmer et al, 2014). The rational for this study was essentially based on the observed clinical responses in a few patients with AML treated in the phase I study (Schimmer et al, 2008). Obatoclax was initially given at 30 mg per day for 3 days, but the dose was reduced to 20 mg per day for 3 days due to neurological toxicity (confusion, somnolence and ataxia) in the first three patients. In the phase II part, patients received either 20 mg per day for 3 days or 60 mg over 24 h. None of the 18 patients who received obatoxclax achieved a CR. From the safety perspective, the most common adverse events were mild transient neurological effects such as euphoria, ataxia, somnolence and dizziness (Schimmer et al, 2014). This adverse effect profile was probably related to a lack of specificity of obatoclax and led to the termination of the programme.
Venetoclax (formerly ABT199), a second-generation, specific antagonist of Bcl-2 showed promising single agent activity in patient-derived AML samples (Pan et al, 2014). These results were confirmed in a recently published study. In this study, venetoclax was administered orally in 32 patients with AML at a dose of 800 mg daily (continuous dosing), with an intrapatient dose escalation during the first week, in order to mitigate the risk of tumour lysis syndrome (Konopleva et al, 2016). The majority of patients had previously received chemotherapy and/or a demethylating agent. The CR/CRi rate was 19% (6 of 32), and another six patients experience bone marrow blast reduction not reaching the threshold for CR (Konopleva et al, 2016). The majority of responses were seen within the first 4 weeks of therapy. However, the duration of complete or partial remission was limited, and all patients discontinued venetoclax due to disease progression. The most common adverse events seen with venetoclax were nausea, diarrhoea, and vomiting as well as febrile neutropenia. In a subsequent study, venetoclax was combined with azacytidine or decitabine in patients with treatment-naïve AML. In this ongoing study, 39 elderly (age⩾65) patients with intermediate or poor-risk cytogenetics not eligible for intensive induction therapy were treated with either decitabine (20 mg m−2, days 1–5) or azacytidine (75 mg m−2 days 1–7) combined with ascending doses of venetoclax daily (Pollyea et al, 2016). Doses of venetoclax up to 800 mg daily were well tolerated and produced a 76% overall response rate, which compares favourably with single agent demethylating agents, and some responses were durable. Based on these promising results a phase 3 trial is currently under way to compare venetoclax+azacytidine to single agent azacytidine in patients with treatment-naïve AML not eligible for intensive therapy (Table 1). In another phase 1b study, the feasibility and efficacy of venetoclax (600 mg per day) combined with low dose cytarabine (20 mg m−2 days 1–10 every 28 days) was assessed in 20 elderly patients with AML, some of them previously treated with demethylating agents. The combination was well tolerated and yielded an overall response rate of 75%. Based on these results a phase 3 trial is currently recruiting to compare venetoclax+low dose cytarabine to low dose cytarabine alone (Table 1). Other BCL2-specific inhibitors, such as S055746 are currently in phase 1 clinical trial (Table 1).
In preclinical models, MCL1 was shown to contribute to resistance to venetoclax in AML cells, and this could be overcome by DNA-damage-mediated reduction in MCL1 levels using standard of care cytotoxics such as daunorubicin and cytarabine (Niu et al, 2016). Although this provides a rational for combining BCL2 inhibitors with standard chemotherapy, overlapping toxicities of venetoclax and cytotoxic agents may limit the feasibility of combining these agents together. Several specific MCL1 inhibitors have recently entered clinical, but no data has yet been reported on their clinical activity (MIK665/S64315-NCT02992483 and NCT02979366; and AMG-176-NCT02675452).
Targeting XIAP and IAP
Several inhibitors of various IAP have entered the clinic in the last decade with a couple being tested in AML or haematology-specific studies. From a pharmacological standpoint the first generation of drugs were in most cases antisense oligonucleotides while later generation agents are more often small molecule SMAC mimetics.
AEG35156 is an antisense oligonucleotide targeting XIAP. The rational for targeting XIAP is based on its potent caspase-inhibitory effect as well as correlative studies implicating XIAP in therapy resistance (Zhang et al, 2002; Tamm et al, 2004). In early phase studies performed in patients with relapsing/refractory patients, interesting results were seen with AEG35156 combined with idarubicin and high dose cytarabine as reinduction: among patients treated at the 350 mg m−2 dose level of AEG35156 15 of 32 (47%) reached a CR/CRp, the CR rate was 10/11 among patients with refractory AML (Schimmer et al, 2009). Based on these promising results, a randomised phase II trial was conducted comparing idarubicin (12 mg m−2 for 3 days) and cytarabine (1.5 g m−2 for 3 or 4 days depending on the patient’s age), with or without AEG35156 650 mg given intravenously on days 1, 2, 3 and 8 (Schimmer et al, 2011). However, the study was terminated early (after 41/60 patients were enrolled) when an interim analysis showed that the study would not meet its primary endpoint of increased CR rate (from 50 to 70%). In the final analysis, the CR/CRp rate was 41% in the experimental arm vs 69% in the control arm (P=0.18). Interestingly, although the addition of AEG35156 did not significantly alter the safety profile of the induction regimen, three patients died during the induction in the AEG35156 arm of the study while none of the 13 patients treated with conventional chemotherapy did (Schimmer et al, 2011).
Erba and colleagues reported on a study investigating LY2181308, a survivin antisense oligonucleotide as a single agent and combined with idarubicin and cytarabine in patients with refractory/relapsed AML (Erba et al, 2013). In both study arms, patients received a loading dose of 750 mg per day of LY2181308 for 3 days, given intravenously, followed by 750 mg every week. In the chemotherapy arm, patients received in addition idarubicin 12 mg m−2 per day and cytarabine 1.5 mg m−2 per day on days 4, 5 and 6. Chemotherapy was repeated every 28 days. Eight patients with high survivin expression (based on flow cytometry analysis at baseline) were treated in the single agent arm and no response was seen although exposure to LY2181308 did reduce survivin in peripheral blasts. In the combination arm, 4/16 (25%) achieved a CR, while a total of 9 patients (56%) were considered to have clinically meaningful response. Moreover, although LY2181308 was responsible for moderate myelosuppression, fatigue and flu-like syndrome (Tanioka et al, 2011) as a single agent, it did not appear to significantly alter the safety of the chemotherapy regimen used in this study (Erba et al, 2013). Although this was a small study with a heterogeneous patient population (all patients were previously treated and 10 patients had had prior transplant), LY2181308 and standard chemotherapy did not appear to lead to increased activity over what is expected from chemotherapy alone. There is currently no ongoing study with LY2181308 (www.clinicaltrials.gov accessed on 13 October 2016).
DiPersio et al recently reported on the activity of Debio-1143 combined with daunorubicin and cytarabine in patients with relapsed or poor risk AML, aged 75 years or less (DiPersio et al, 2015). Debio-1143 is an orally administered, monovalent SMAC-mimetic and binds preferentially to cIAP1 and cIAP2, and with less affinity to XIAP (Cai et al, 2011). The maximum tolerated dose of Debio-1143, administered orally on days 1–5, combined with the standard ‘7 plus 3 regimen’ was 400 mg given once daily. Although doses of 200 and 300 mg were shown to be safe, the 100 mg dose was selected for further studies due to apparent increased efficacy in the 100 mg cohort. A total of 11 patients achieved a CR, two patients had a CRi, while one patient had a partial response (overall response rate 14/29, 48%)(DiPersio et al, 2015). The most common adverse events related to treatment were nausea, diarrhoea and febrile neutropenia which is consistent with some of the side effects seen with single agent Debio1143 (nausea, vomiting and fatigue)(Hurwitz et al, 2015).
Birinapant (formerly TL32711) is a bivalent SMAC-mimetic that displays preferential binding to cIAP1 relative to cIAP2 and XIAP (Condon et al, 2014). In preclinical studies, birinapant showed potent anti-tumour activity alone and combined with various agents, including azacitidine, a demethylating agent, in models of AML (Carter et al, 2014). Preliminary data from a phase 1, dose and schedule optimisation study of birinapant were reported at the 2014 annual meeting of American Society of Hematology. In this study patients 20 with AML (n=19) and myelodysplastic syndrome (MDS) (n=1) were treated at various dose levels and schedules. Birinapant 17 mg m−2 twice a week appeared to be the optimal schedule in this early report. The best response to therapy was stable disease observed in some patients (Frey et al, 2014). The most common adverse events seen with birinapant include nausea, vomiting and diarrhoea, as well as fatigue, fever and cytokine release syndrome, which seems to be an on-target toxicity of IAP inhibitors. Bell’s palsy was also a dose-limiting toxicity with birinapant and was observed in patients with AML but also those with solid tumours treated in a separate study (Amaravadi et al, 2015), as well as those treated with other bivalent IAP inhibitors (Sikic et al, 2011), which suggests that it may be an on-target effect, although the mechanism remains uncertain. A study combining birinapant with azacitidine in patients with MDS and chronic myelomonogenous leukaemia was initiated based on the data reported by Carter et al (2014). However, this study was prematurely terminated when a preplanned interim analysis showed no increased efficacy for azacitidine+birinapant vs azacytidine alone (Donnellan et al, 2016). Interestingly, Brumatti et al recently showed that co-administration of a caspase inhibitor may increase birinapant-induced cell death by blocking caspase-8 and apoptosis, and inducing necroptosis (Brumatti et al, 2016). This opens the way for therapeutically targeting alternative cell death mechanisms such as necroptosis. The same group reported on the synergistic effect of combining a p38α inhibitor (such as LY2228820) with birinapant on AML both in vitro and in vivo, where the effect of the combination was this time dependent on TNF-induced apoptosis, and not necroptosis (Lalaoui et al, 2016).
Data regarding another inhibitor of SMAC mimetic (CUDC-427) were recently published (Tolcher et al, 2016), but no safety or efficacy data have yet been reported in patients with haematological malignancies. Likewise, initial safety data were reported for LCL161, another SMAC mimetic, as well as interesting data in combination with cyclophosphamide in patients with multiple myeloma but there is no ongoing study in patients with AML (Infante et al, 2014; Chesi et al, 2016).
Targeting death receptors
Although death receptors are expressed in AML cells and some preclinical data indicate in vitro activity of DR4 agonist, there has been no clinical development of these drugs in AML, likely because of their collective lack of clinical activity in solid tumours. Newer compounds are currently being evaluated such as bispecific antibodies, but their relevance to AML is currently unknown (e.g. RO6874813, currently in phase 1 clinical trial). The recent observation that DR5 agonist may be able to downregulate myeloid-derived suppressor cells in vivo (Dominguez et al, 2017) may renew interest in death receptor agonists as potential combination partners with other immune modulating agents. Similarly, Chesi and colleagues recently showed that aside from their effect as inducers of apoptosis, SMAC mimetic (IAP antagonists) were able to induce anti-tumour immunity by modulating the NF-κB response in immune cells in the tumour microenvironment (Chesi et al, 2016). Together these data suggest that because the extrinsic apoptotic pathway regulates both apoptosis and the inflammatory response through NF-κB, drugs targeting this pathway (namely death receptor agonists and SMAC mimetics) may have immune modulatory properties that may allow their combination with other immune modulatory agents such as inhibitors of the immune checkpoints.
Conclusion
Targeting the apoptosis machinery is a promising therapeutic approach in AML. In most cases, the goal is to eliminate leukaemic cells by reactivating cell death pathways, either by direct targeting of anti-apoptotic proteins such as those of the Bcl-2 or the IAP families, or by reactivating the p53 response. Most of the molecules targeting anti-apoptotic proteins that entered clinical trials demonstrated their tolerability, at least when tested alone, and in some cases when used in combination with cytotoxics. The full therapeutic activity of these molecules will probably be best realised through combination with other, cytotoxic or targeted, anticancer agents. Demethylating agents for example may be good mechanistic candidates for such approaches, because they induce the expression of death receptors in leukaemic cells (Karlic et al, 2014) but also sensitise leukaemic cells to IAP inhibitors (Carter et al, 2014), although the clinical relevance of this finding still remains to be shown.
Combinations of different drugs targeting apoptosis can also be envisaged. A recent study showed that the combination of an anti-MDM2 and a BET-inhibitor (targeting p53 and Myc) was able to eliminate leukaemic stem cells in a chronic myelogenous leukaemia model (Abraham et al, 2016). Other combinations are currently under clinical evaluation, including Bcl-2 inhibitors combined with MDM2 antagonists (Lehmann et al, 2016) or MEK inhibitors (Zhang et al, 2010) (Table 1). Apoptosis-inducing agents such as MDM2 or Bcl-2 antagonists may then represent an ‘apoptosis backbone’ for combination therapies in AML; however, the optimal combination partners remain to be determined. In their recently reported study, Gu and colleagues showed interesting activity of new MDM2 inhibitors that compromise XIAP mRNA translation by targeting the MDM2 RING domain, in an acute lymphoblastic leukaemia model (Gu et al, 2016). Taken together, these reports suggest that targeting simultaneously several nodes of apoptosis regulation may be more efficacious than targeting a single node. Also, emerging data suggest that alterations implicating alternative cell death mechanisms, such as autophagy and necroptosis for example, play a role in oncogenesis and tumour progression in various cancer models, including leukaemia. For example, autophagy which is an adaptive survival mechanism that allows the recycling of cellular constituents during conditions of cellular stress has been shown to contribute to leukaemia initiation and resistance to therapy, although this seems to be subtype specific (Auberger and Puissant, 2017). Similarly, necroptosis is a recently identified mechanism of regulated cell death.
One key question is the integration of these promising agents in the current therapeutic armamentarium. The only agent currently in late stage investigation is the MDM2 inhibitor idasanutlin (NCT02545283). This study compares cytarabine alone to cytarabine combined with idasanutlin, in first or second relapse, after failure of at least one conventional intensive induction regimen. Overall some overlapping toxicities of apoptosis targeting drugs with those of conventional cytotoxic agents may render their combination quite toxic or feasible only in young and fit patients. The feasibility of combining apoptosis targeting drugs with each other or with other targeted agents remains to be demonstrated in the clinic as is their relevance to clinical subgroups of AML patients. Indeed, many patients with AML may not be candidate to intensive induction chemotherapy because of older age or poor risk features. Overall, three settings can be envisioned: (1) combination with standard of care chemotherapy to improve percentage and/or duration or response for fit patients; (2) treatment of fit patients with chemotherapy-refractory disease and/or for patients with poor risk cytogenetics; this will probably involve combinations of apoptosis-targeting drugs with other targeted agents (demethylating agents for example); and (3) therapy for unfit patient, which will likely involve single agent regimens or carefully selected combinations.
The field of AML research is currently being revolutionised by the recently published molecular classifications (Patel et al, 2012; Cancer Genome Atlas Research Network, 2013; Papaemmanuil et al, 2016). It is tempting to speculate that these classifications may ultimately guide therapy for patients with AML, in a so-called ‘precision medicine’ approach. Recent examples including the development of FLT3 or IDH1-2 inhibitors illustrate this concept (Cortes et al, 2013; Hansen et al, 2014; Stein et al, 2014). However, a large proportion of patients harbour non-druggable genomic alterations. There is thus room for alternative strategies like the targeting of apoptosis that should be active across molecular classes of the disease. How molecular alterations will correlate with clinical activity of modulators of apoptosis is currently largely unknown. Of course, mutations in the TP53 gene have been shown as a mechanism of both primary and secondary resistance to MDM2 inhibitors (Jeay et al, 2015), but the molecular alterations underpinning the activity of other inhibitors of anti-apoptotic proteins in AML remains to be identified. Currently, the expression of Bcl-2, MDM2 and XIAP seems to be the biomarker of choice for Bcl-2 inhibitors, MDM2 antagonists and XIAP inhibitors/SMAC-mimetics, but the expression of these proteins may vary at different stages of the disease (primary diagnosis vs relapse vs refractory setting) and how this correlates with the various subclasses of AML is still currently under investigation.
References
Abraham SA, Hopcroft LE, Carrick E, Drotar ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT, Pellicano F, Pierce A, Copland M, Nourse C, Grimmond SM, Vetrie D, Whetton AD, Holyoake TL (2016) Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 534 (7607): 341–346.
Amaravadi RK, Schilder RJ, Martin LP, Levin M, Graham MA, Weng DE, Adjei AA (2015) A Phase I study of the SMAC-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol Cancer Ther 14 (11): 2569–2575.
Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, Kirschbaum M, Iyer SP, Ruvolo V, González GM, Huang X, Chen G, Graves B, Blotner S, Bridge P, Jukofsky L, Middleton S, Reckner M, Rueger R, Zhi J, Nichols G, Kojima K (2016) Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res 22 (4): 868–876.
Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ (2017) From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov 16 (4): 273–284.
Auberger P, Puissant A (2017) Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 129 (5): 547–552.
Brumatti G, Ma C, Lalaoui N, Nguyen NY, Navarro M, Tanzer MC, Richmond J, Ghisi M, Salmon JM, Silke N, Pomilio G, Glaser SP, de Valle E, Gugasyan R, Gurthridge MA, Condon SM, Johnstone RW, Lock R, Salvesen G, Wei A, Vaux DL, Ekert PG, Silke J (2016) The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci Transl Med 8 (339): 339ra69.
Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang CY, Miller R, Yi H, Zhang T, Sun D, Kang S, Guo M, Leopold L, Yang D, Wang S (2011) A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem 54 (8): 2714–2726.
Cancer Genome Atlas Research Network (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368 (22): 2059–2074.
Carter BZ, Mak PY, Mak DH, Shi Y, Qiu Y, Bogenberger JM, Mu H, Tibes R, Yao H, Coombes KR, Jacamo RO, McQueen T, Kornblau SM, Andreeff M (2014) Synergistic targeting of AML stem/progenitor cells with IAP antagonist birinapant and demethylating agents. J Natl Cancer Inst 106 (2): djt440.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2 (5): 401–404.
Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, Akhmetzyanova I, Kosiorek HE, Calcinotto A, Riggs DL, Keane N, Ahmann GJ, Morrison KM, Fonseca R, Lacy MQ, Dingli D, Kumar SK, Ailawadhi S, Dispenzieri A, Buadi F, Gertz MA, Reeder CB, Lin Y, Chanan-Khan AA, Stewart AK, Fooksman D, Bergsagel PL (2016) IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med 22 (12): 1411–1420.
Condon SM, Mitsuuchi Y, Deng Y, LaPorte MG, Rippin SR, Haimowitz T, Alexander MD, Kumar PT, Hendi MS, Lee YH, Benetatos CA, Yu G, Kapoor GS, Neiman E, Seipel ME, Burns JM, Graham MA, McKinlay MA, Li X, Wang J, Shi Y, Feltham R, Bettjeman B, Cumming MH, Vince JE, Khan N, Silke J, Day CL, Chunduru SK (2014) Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J Med Chem 57 (9): 3666–3677.
Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, Gammon G, Trone D, Armstrong RC, James J, Levis M (2013) Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol 31 (29): 3681–3687.
Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S (2006) Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem 49 (12): 3432–3435.
DiPersio JF, Erba HP, Larson RA, Luger SM, Tallman MS, Brill JM, Vuagniaux G, Rouits E, Sorensen JM, Zanna C (2015) Oral Debio1143 (AT406), an antagonist of inhibitor of apoptosis proteins, combined with daunorubicin and cytarabine in patients with poor-risk acute myeloid leukemia – results of a phase I dose-escalation study. Clin Lymphoma Myeloma Leuk 15 (7): 443–449.
Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B, Bloomfield CD European LeukemiaNet (2010) Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115 (3): 453–474.
Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, Forero A, Bendell J, Witt R, Hockstein N, Kumar P, Gabrilovich DI (2017) Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res 23 (12): 2942–2950.
Donnellan WB, Diez-Campelo M, Heuser M, Ritchie EK, Skolnik J, Font P, Komrokji RS (2016) A phase 2 study of azacitidine (5-AZA) with or without birinapant in subjects with higher risk myelodysplastic syndrome (MDS) or chronic myelomonocytic leukemia (CMML). J Clin Oncol 34 (suppl; abstr 7060).
Erba HP, Sayar H, Juckett M, Lahn M, Andre V, Callies S, Schmidt S, Kadam S, Brandt JT, Van Bockstaele D, Andreeff M (2013) Safety and pharmacokinetics of the antisense oligonucleotide (ASO) LY2181308 as a single-agent or in combination with idarubicin and cytarabine in patients with refractory or relapsed acute myeloid leukemia (AML). Invest New Drugs 31 (4): 1023–1034.
Ettou S, Humbrecht C, Benet B, Billot K, d’Allard D, Mariot V, Goodhardt M, Kosmider O, Mayeux P, Solary E, Fontenay M (2013) Epigenetic control of NF-κB-dependent FAS gene transcription during progression of myelodysplastic syndromes. Mol Cancer Res 11 (7): 724–735.
Faderl S, Kantarjian HM, Estey E, Manshouri T, Chan CY, Rahman Elsaied A, Kornblau SM, Cortes J, Thomas DA, Pierce S, Keating MJ, Estrov Z, Albitar M (2000) The prognostic significance of p16(INK4a)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 89 (9): 1976–1982.
Fathi AT, Abdel-Wahab O (2012) Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Adv Hematol 2012: 469592.
Fesik SW (2005) Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer 5 (11): 876–885, (review). Erratum in: Nat Rev Cancer 2005;5(12):995.
Ferrara F, Schiffer CA (2013) Acute myeloid leukaemia in adults. Lancet 381 (9865): 484–495.
Frey NV, Luger S, Mangan J, Zebrowski A, Loren AW, Minderman H, Baird J, Porter DL, Hexner EO, Kumar AJ, Luskin MR, Perl AE, Carroll M (2014) A phase I study using single agent birinapant in patients with relapsed myelodysplastic syndrome and acute myelogenous leukemia. Blood 124: 3758.
Fukuhara S, Rowley JD (1978) Chromosome 14 translocations in non-Burkitt lymphomas. Int J Cancer 22 (1): 14–21.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6 (269): pl1.
Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, Alexander WS, Lowe SW, Robb L, Strasser A (2012) Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev 26 (2): 120–125.
Gu L, Zhang H, Liu T, Zhou S, Du Y, Xiong J, Yi S, Qu CK, Fu H, Zhou M (2016) Discovery of dual inhibitors of MDM2 and XIAP for cancer treatment. Cancer Cell 30 (4): 623–636.
Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T (2008) Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 22 (8): 1539–1541.
Hainaut P, Pfeifer GP (2016) Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med 6 (11):: pii: a026179.
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5): 646–674.
Hansen E, Quivoron C, Straley K, Lemieux RM, Popovici-Muller J, Sadrzadeh H, Fathi AT, Gliser C, David M, Saada V, Micol J, Bernard O, Dorsch M, Yang H, Su M, Agresta S, de Botton S, Penard-Lacronique V, Yen K (2014) AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated ex vivo. Blood 124 (21): 3734.
Hata AN, Engelman JA, Faber AC (2015) The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov 5 (5): 475–487.
Hurwitz HI, Smith DC, Pitot HC, Brill JM, Chugh R, Rouits E, Rubin J, Strickler J, Vuagniaux G, Sorensen JM, Zanna C (2015) Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: results of a first-in-man study. Cancer Chemother Pharmacol 75 (4): 851–859.
Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, Cohen RB (2014) Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol 32 (28): 3103–3110.
Jeay S, Gaulis S, Ferretti S, Bitter H, Ito M, Valat T, Murakami M, Ruetz S, Guthy DA, Rynn C, Jensen MR, Wiesmann M, Kallen J, Furet P, Gessier F, Holzer P, Masuya K, Würthner J, Halilovic E, Hofmann F, Sellers WR, Graus Porta D (2015) A distinct p53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. Elife 4.
Jung J, Lee JS, Dickson MA, Schwartz GK, Le Cesne A, Varga A, Bahleda R, Wagner AJ, Choy E, de Jonge MJ, Light M, Rowley S, Macé S, Watters J (2016) TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun 7: 12609.
Karlic H, Herrmann H, Varga F, Thaler R, Reitermaier R, Spitzer S, Ghanim V, Blatt K, Sperr WR, Valent P, Pfeilstöcker M (2014) The role of epigenetics in the regulation of apoptosis in myelodysplastic syndromes and acute myeloid leukemia. Crit Rev Oncol Hematol 90 (1): 1–16.
Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, Blum W, DiNardo CD, Kadia T, Dunbar M, Kirby R, Falotico N, Leverson J, Humerickhouse R, Mabry M, Stone R, Kantarjian H, Letai A (2016) Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 6 (10): 1106–1117.
Kornblau SM, Thall PF, Estrov Z, Walterscheid M, Patel S, Theriault A, Keating MJ, Kantarjian H, Estey E, Andreeff M (1999) The prognostic impact of BCL2 protein expression in acute myelogenous leukemia varies with cytogenetics. Clin Cancer Res 5 (7): 1758–1766.
Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, Bruno A, Csekei M, Paczal A, Szabo ZB, Sipos S, Radics G, Proszenyak A, Balint B, Ondi L, Blasko G, Robertson A, Surgenor A, Dokurno P, Chen I, Matassova N, Smith J, Pedder C, Graham C, Studeny A, Lysiak-Auvity G, Girard AM, Gravé F, Segal D, Riffkin CD, Pomilio G, Galbraith LC, Aubrey BJ, Brennan MS, Herold MJ, Chang C, Guasconi G, Cauquil N, Melchiore F, Guigal-Stephan N, Lockhart B, Colland F, Hickman JA, Roberts AW, Huang DC, Wei AH, Strasser A, Lessene G, Geneste O (2016) The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538 (7626): 477–482.
Kurzrock K, Blay JY, Bui BN, Wagner AJ, Maki RG, Schwartz GK, Patnaik A, Gore L, Wu L, Vassilev LT, Ding M, Geho D, Zhi J, Middleton S, Nichols GL (2012) A phase I study of MDM2 antagonist RG7112 in patients (pts) with relapsed/refractory solid tumors. J Clin Oncol 30 (suppl; abstr e13600).
Lalaoui N, Hänggi K, Brumatti G, Chau D, Nguyen NN, Vasilikos L, Spilgies LM, Heckmann DA, Ma C, Ghisi M, Salmon JM, Matthews GM, de Valle E, Moujalled DM, Menon MB, Spall SK, Glaser SP, Richmond J, Lock RB, Condon SM, Gugasyan R, Gaestel M, Guthridge M, Johnstone RW, Munoz L, Wei A, Ekert PG, Vaux DL, Wong WW, Silke J (2016) Targeting p38 or MK2 enhances the anti-leukemic activity of Smac-mimetics. Cancer Cell 30 (3): 499–500.
Lehmann C, Friess T, Birzele F, Kiialainen A, Dangl M (2016) Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J Hematol Oncol 9 (1): 50.
Letai AG (2008) Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer 8 (2): 121–132.
Long J, Parkin B, Ouillette P, Bixby D, Shedden K, Erba H, Wang S, Malek SN (2010) Multiple distinct molecular mechanisms influence sensitivity and resistance to MDM2 inhibitors in adult acute myelogenous leukemia. Blood 116 (1): 71–80.
Mehta SV, Shukla SN, Vora HH (2013) Overexpression of Bcl2 protein predicts chemoresistance in acute myeloid leukemia: its correlation with FLT3. Neoplasma 60 (6): 666–675.
Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, Caldwell JT, Xiang S, Zhang X, Chu R, Wang ZJ, Lin H, Taub JW, Ge Y (2016) Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res 22 (17): 4440–4451.
Obexer P, Ausserlechner MJ (2014) X-linked inhibitor of apoptosis protein – a critical death resistance regulator and therapeutic target for personalized cancer therapy. Front Oncol 4: 197.
Ossenkoppele G, Löwenberg B (2015) How I treat the older patient with acute myeloid leukemia. Blood 125 (5): 767–774.
Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, Döhner H, Gaidzik VI, Galinsky I, Golfman LS, Haferlach T, Harutyunyan KG, Hu J, Leverson JD, Marcucci G, Müschen M, Newman R, Park E, Ruvolo PP, Ruvolo V, Ryan J, Schindela S, Zweidler-McKay P, Stone RM, Kantarjian H, Andreeff M, Konopleva M, Letai AG (2014) Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 4 (3): 362–375.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, Gundem G, Van Loo P, Martincorena I, Ganly P, Mudie L, McLaren S, O’Meara S, Raine K, Jones DR, Teague JW, Butler AP, Greaves MF, Ganser A, Döhner K, Schlenk RF, Döhner H, Campbell PJ (2016) Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 374 (23): 2209–2221.
Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, Vance G, Higgins RR, Ketterling RP, Gallagher RE, Litzow M, van den Brink MR, Lazarus HM, Rowe JM, Luger S, Ferrando A, Paietta E, Tallman MS, Melnick A, Abdel-Wahab O, Levine RL (2012) Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 366 (12): 1079–1089.
Phillips A, Teunisse A, Lam S, Lodder K, Darley M, Emaduddin M, Wolf A, Richter J, de Lange J, Verlaan-de Vries M, Lenos K, Böhnke A, Bartel F, Blaydes JP, Jochemsen AG (2010) HDMX-L is expressed from a functional p53-responsive promoter in the first intron of the HDMX gene and participates in an autoregulatory feedback loop to control p53 activity. J Biol Chem 285 (38): 29111–29127.
Pluta A, Wierzbowska A, Cebula-Obrzut B, Pluta P, Stępka K, Szmigielska-Kapłon A, Grzybowska-Izydorczyk O, Czemerska M, Smolewski P, Wrzesien-Kus A, Robak T (2015) Prognostic value of inhibitor of apoptosis protein family expression in patients with acute myeloid leukemia. Leuk Lymphoma 56 (9): 2529–2535.
Pollyea DA, Dinardo CD, Thirman MJ, Letai A, Wei AH, Jonas BA, Arellano ML, Frattini MG, Kantarjian HM, Chyla B, Zhu M, Potluri J, Humerickhouse R, Mabry MH, Konopleva M, Pratz KW (2016) Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ⩾ 65 years ineligible for standard induction therapy. In: Proceedings of the ASCO Meeting 2016.
Quintás-Cardama A, Hu C, Qutub A, Qiu YH, Zhang X, Post SM, Zhang N, Coombes K, Kornblau SM (2017) p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 31 (6): 1296–1305.
Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO, Fancourt C, Lam R, Jones MB, Knox CD, Rose S, Patel PS, Tibes R (2016) A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk Res 48: 92–100.
Reis B, Jukofsky L, Chen G, Martinelli G, Zhong H, So WV, Dickinson MJ, Drummond M, Assouline S, Hashemyan M, Theron M, Blotner S, Lee JH, Kasner M, Yoon SS, Rueger R, Seiter K, Middleton SA, Kelly KR, Vey N, Yee K, Nichols G, Chen LC, Pierceall WE (2016) Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica 101 (5): e185–e188.
Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI, Paschka P, Held G, von Lilienfeld-Toal M, Lübbert M, Fröhling S, Zenz T, Krauter J, Schlegelberger B, Ganser A, Lichter P, Döhner K, Döhner H (2012) TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 119 (9): 2114–2121.
Schimmer AD, O’Brien S, Kantarjian H, Brandwein J, Cheson BD, Minden MD, Yee K, Ravandi F, Giles F, Schuh A, Gupta V, Andreeff M, Koller C, Chang H, Kamel-Reid S, Berger M, Viallet J, Borthakur G (2008) A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin Cancer Res 14 (24): 8295–8301.
Schimmer AD, Estey EH, Borthakur G, Carter BZ, Schiller GJ, Tallman MS, Altman JK, Karp JE, Kassis J, Hedley DW, Brandwein J, Xu W, Mak DH, LaCasse E, Jacob C, Morris SJ, Jolivet J, Andreeff M (2009) Phase I/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J Clin Oncol 27 (28): 4741–4746.
Schimmer AD, Herr W, Hanel M, Borthakur G, Frankel A, Horst HA, Martin S, Kassis J, Desjardins P, Seiter K, Fiedler W, Noppeney R, Giagounidis A, Jacob C, Jolivet J, Tallman MS, Koschmieder S (2011) Addition of AEG35156 XIAP antisense oligonucleotide in reinduction chemotherapy does not improve remission rates in patients with primary refractory acute myeloid leukemia in a randomized phase II study. Clin Lymphoma Myeloma Leuk 11 (5): 433–438.
Schimmer AD, Raza A, Carter TH, Claxton D, Erba H, DeAngelo DJ, Tallman MS, Goard C, Borthakur G (2014) A multicenter phase I/II study of obatoclax mesylate administered as a 3- or 24-hour infusion in older patients with previously untreated acute myeloid leukemia. PLoS One 9 (10): e108694.
Schlereth K, Beinoraviciute-Kellner R, Zeitlinger MK, Bretz AC, Sauer M, Charles JP, Vogiatzi F, Leich E, Samans B, Eilers M, Kisker C, Rosenwald A, Stiewe T (2010) DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 38 (3): 356–368.
Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, Antony-Debre I, Viale A, Socci N, Sun Y, Robertson A, Cavatore M, de Stanchina E, Hricik T, Rapaport F, Woods B, Wei C, Hatlen M, Baljevic M, Nimer SD, Tallman M, Paietta E, Cimmino L, Aifantis I, Steidl U, Mason C, Melnick A, Levine RL (2015) Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 27 (4): 502–515.
Sikic BI, Eckhardt SG, Gallant GJA, Burris HA, Camidge DR, Colevas AD, Jones SF, Messersmith WA, Wakelee HA, Li H, Kaminker PG, Morris S, Infante JR (2011) Safety, pharmacokinetics (PK), and pharmacodynamics (PD) of HGS1029, an inhibitor of apoptosis protein (IAP) inhibitor, in patients (Pts) with advanced solid tumors: Results of a phase I study. J Clin Oncol 29 (suppl; abstr 3008).
Soderquist R, Eastman A (2016) BCL2 inhibitors as anticancer drugs: a plethora of misleading BH3 mimetics. Mol Cancer Ther 15 (9): 2011–2017.
Stein EM, Altman JK, Collins R, DeAngelo DJ, Fathi AT, Flinn I, Frankel A, Levine RL, Medeiros BC, Patel M, Pollyea DA, Roboz GJ, Stone RM, Swords RT, Tallman MS, Agresta S, FanB, Yang H, Yen K, de Botton S (2014) AG-221, an oral, selective, first-in-class, potent inhibitor of the IDH2 mutant metabolic enzyme, induces durable remissions in a phase I study in patients with IDH2 mutation positive advanced hematologic malignancies. Blood 124 (21): 115.
Siu LS, Italiano A, Miller WH, Blay J-Y, Gietema JA, Bang Y-J, Mileshkin LR, Hirte HW, Reckner M, Higgins B, Jukofsky L, Blotner S, Zhi J, Middleton S, Nichols GL, Chen LC (2014) Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist, in patients (pts) with solid tumors. J Clin Oncol 32: 5s (suppl; abstr 2535).
Tamm I, Richter S, Oltersdorf D, Creutzig U, Harbott J, Scholz F, Karawajew L, Ludwig WD, Wuchter C (2004) High expression levels of x-linked inhibitor of apoptosis protein and survivin correlate with poor overall survival in childhood de novo acute myeloid leukemia. Clin Cancer Res 10 (11): 3737–3744.
Tanioka M, Nokihara H, Yamamoto N, Yamada Y, Yamada K, Goto Y, Fujimoto T, Sekiguchi R, Uenaka K, Callies S, Tamura T (2011) Phase I study of LY2181308, an antisense oligonucleotide against survivin, in patients with advanced solid tumors. Cancer Chemother Pharmacol 68 (2): 505–511.
Tolcher AW, Bendell JC, Papadopoulos KP, Burris HA, Patnaik A, Fairbrother WJ, Wong H, Budha N, Darbonne WC, Peale F, Mamounas M, Royer-Joo S, Yu R, Portera CC, Infante JR (2016) A phase I dose-escalation study evaluating the safety tolerability and pharmacokinetics of CUDC-427, a potent, oral, monovalent IAP antagonist, in patients with refractory solid tumors. Clin Cancer Res 22 (18): 4567–4573.
Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM (1984) Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science 224 (4656): 1403–1406.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC (2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10 (5): 389–399.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303 (5659): 844–848.
Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, Adams JM, Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302 (5647): 1036–1038.
Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch HA, Fancour C, Levonas A, Lam R, Meister A, Kemp RK, Knox C, Rose S, Hong DS (2015) A phase I trial of the human double minute 2 (HDM2) inhibitor MK-8242 in patients (pts) with advanced solid tumors. J Clin Oncol 33 (12): 1304–1311, (suppl; abstr 10564).
Weisberg E, Halilovic E, Cooke VG, Nonami A, Ren T, Sanda T, Simkin I, Yuan J, Antonakos B, Barys L, Ito M, Stone R, Galinsky I, Cowens K, Nelson E, Sattler M, Jeay S, Wuerthner JU, McDonough SM, Wiesmann M, Griffin JD (2015) Inhibition of wild-type p53-expressing AML by the novel small molecule HDM2 inhibitor CGM097. Mol Cancer Ther 14 (10): 2249–2259.
Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, Wilson RK, Baty JD, Duncavage EJ, Tandon B, Lee YS, Wartman LD, Uy GL, Ghobadi A, Tomasson MH, Pusic I, Romee R, Fehniger TA, Stockerl-Goldstein KE, Vij R, Oh ST, Abboud CN, Cashen AF, Schroeder MA, Jacoby MA, Heath SE, Luber K, Janke MR, Hantel A, Khan N, Sukhanova MJ, Knoebel RW, Stock W, Graubert TA, Walter MJ, Westervelt P, Link DC, DiPersio JF, Ley TJ (2016) TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 375 (21): 2023–2036.
Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, Heath S, Baty JD, Klco JM, Ding L, Mardis ER, Westervelt P, DiPersio JF, Walter MJ, Graubert TA, Ley TJ, Druley TE, Link DC, Wilson RK (2015) Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 518 (7540): 552–555.
Xiang Z, Luo H, Payton JE, Cain J, Ley TJ, Opferman JT, Tomasson MH (2010) Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest 120 (6): 2109–2118.
Yee K, Martinelli G, Vey N, Dickinson MJ, Seiter K, Assouline S, Drummond M, Yoon SS, Kasner M, Lee JH, Kelly KR, Blotner S, Higgins B, Middleton S, Nichols G, Chen G, Zhong H, Pierceall WE, Zhi J, Chen LC (2014) Phase 1/1b study of RG7388, a potent MDM2 antagonist, in acute myelogenous leukemia (AML) patients (Pts). Blood 124 (21): 116–116.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7 (3): 673–682.
Zhang J, Li Y, Shen B (2002) Up-regulation of XIAP by M-CSF is associated with resistance of myeloid leukemia cells to apoptosis. Leukemia 16 (10): 2163–2165.
Zhang W, Konopleva M, Burks JK, Dywer KC, Schober WD, Yang JY, McQueen TJ, Hung MC, Andreeff M (2010) Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase and murine double minute synergistically induces Apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res 70 (6): 2424–2434.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
PAC has received honorarium and/or research funding from Amgen, Astra-Zeneca, Bayer, Blueprint Medicines, Bristol Myers Squib, Celgene, Lilly, Merck Serono, Merck Sharp Dohme, Novartis, Roche/Genentech. AB has received honorarium and/or institutional research funding from Celgene, Novartis, Bristol Myers Squib, Pfizer. NV has received honorarium from Amgen, Celgene, Novartis, Servier. MC declares no relevant conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Cassier, P., Castets, M., Belhabri, A. et al. Targeting apoptosis in acute myeloid leukaemia. Br J Cancer 117, 1089–1098 (2017). https://doi.org/10.1038/bjc.2017.281
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.281
Keywords
This article is cited by
-
Venetoclax therapy and emerging resistance mechanisms in acute myeloid leukaemia
Cell Death & Disease (2024)
-
A biochemical assessment of apoptosis-inducing impact of Salinomycin in combination with ciprofloxacin on human leukemia KG1–a stem-like cells in the presence and absence of insulin
Molecular Biology Reports (2024)
-
STYXL1 promotes proliferation and epithelial mesenchymal transition of gastric cancer cells via activating the PI3K/AKT pathway
Molecular & Cellular Toxicology (2024)
-
Sennoside A induces autophagic death of prostate cancer via inactivation of PI3K/AKT/mTOR axis
Journal of Molecular Histology (2023)
-
The effect of apigenin and chemotherapy combination treatments on apoptosis-related genes and proteins in acute leukaemia cell lines
Scientific Reports (2022)
