
Abstract
An early intervention using biomarkers to predict acute myocardial infarction (AMI) will effectively reduce global heart attack incidence, particularly among high-risk patients with type 2 diabetes mellitus (T2DM). This study attempted to identify potential biomarkers by detecting changes in the levels of plasma proteins in T2DM patients following onset of AMI in comparison with those without AMI. Volunteer T2DM patients without AMI (control; n=10) and T2DM patients with AMI (n=10) were recruited. Plasma samples from these patients were evaluated via two-dimensional gel electrophoresis (2DE) to screen for proteins with level changes between the two groups. The abundance of spots on gel images was analyzed using Progenesis SameSpots and subjected to false discovery rate (FDR) analysis. Protein spots with statistically significant changes of at least 1.5 fold were selected for mass spectrometry (MS) analysis. Due to strong cardiac connections, tetranectin and titin were evaluated by enzymelinked immunosorbent assay (ELISA). The adjusted P-values and fold changes between the two groups resulted in identification of 34 protein spots with significantly altered abundance. Upon MS analysis, 17 plasma proteins were identified: tetranectin, titin, clusterin, haptoglobin, myosin-13, zinc fnger protein 445, DNA repair protein RAD50, serum albumin, apolipoprotein A-IV, caspase-6, aminoacyl tRNA synthase complex-interacting multifunctional protein 1, serotransferrin, retinol-binding protein 4, transthyretin, alpha-1-antitrypsin, apolipoprotein A-I and serum amyloid A. Comparable patterns of changes in tetranectin and titin between the control and AMI groups were confirmed using ELISA. In summary, tetranectin and titin in plasma appeared to be closely associated with the onset of AMI among T2DM patients and can be used as potential biomarkers for prediction of a cardiac event, though this requires validation in a prospective cohort study.
Similar content being viewed by others

Introduction
Acute myocardial infarction (AMI) was responsible for the deaths of at least seven million patients in 20121. According to a statistical analysis, type 2 diabetes mellitus (T2DM) is an inflammatory illness with a clear-cut relationship with cardiovascular disease (CVD) (American Heart Association, 2017; http://www.heart.org/HEARTORG/Conditions/Diabetes/WhyDiabetesMatters/Cardiovascular-Disease-Diabetes_UCM_313865_Article.jsp). In one study, adults with T2DM were shown to have a two to four times higher risk of CVD compared with adults without the disorder and were a high-risk group for AMI2. It is estimated that by the year 2035, 592 million people globally, predominantly in developing countries, will have metabolic syndrome3, which is linked with the cardiac condition.
Through proteomics, many researchers have been studying the use of biomarkers due to their ability to monitor susceptibility to as well as progression and resolution of diseases, health conditions, treatment outcomes, and post-mortem analysis4. Proteomics has been widely used as a means of identifying candidate biomarkers and immunochemistry markers for various illnesses, particularly infectious and neoplastic diseases5. In light of this approach, the Human Proteome Project aims to map all human proteins by characterizing all 20 300 genes of the known genome6. Together with the availability of the entire human coding sequence in the Human Genome Project, the proteome project marks the beginning of a new era for generating a deeper understanding of both human genes and proteins.
In a proteomics workflow, two-dimensional gel electrophoresis (2DE) delivers a map of intact proteins that reflects changes in the protein abundance level, isoforms, or post-translational modifications (PTMs) based on the isoelectric point, relative molecular mass, solubility, and relative abundance of proteins7. These properties notwithstanding, 2DE allows for the isolation of proteins in microgram amounts for further structural analysis by mass spectrometry (MS) or protein sequencing. MS has assumed a key role for identification and precise quantification of proteins from complex samples, thereby enabling large-scale and high-throughput characterization of the human proteome8,9. Because of its simplicity, accuracy, high resolution and sensitivity, matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) is useful for the identification of proteins by peptide mass fingerprinting.
Clinically, human serum proteins have been extensively used as biomarkers for human disease detection, including in various cancers, neurodegenerative diseases, and hepatic cirrhosis. Because human plasma contains an enormously complex mixture of over 700 proteins, including clotting factors, plasma has recently been further investigated and has shown potential for diagnosing Alzheimer's and atherosclerotic diseases10,11. In a clinical setting, natriuretic peptides, C-reactive protein, creatine kinase, and cardiac troponin are already used as biomarkers in acute cardiac care12. Therefore, the human plasma proteome has the potential to predict the onset of AMI. Because acute AMI contributes to a large percentage of mortality worldwide, early intervention using biomarkers that can predict AMI, particularly in high-risk T2DM patients, may help reduce the global heart attack incidence.
In this study, plasma samples were collected from T2DM volunteers with AMI three days after admission, and T2DM subjects without AMI were the control group. Both samples were subjected to 2DE, and proteins were separated according to their isoelectric points and relative molecular masses. Proteins with changes in abundance were subsequently identified using MALDI-TOF/TOF analysis and quantitatively validated with enzyme-linked immunosorbent assay (ELISA). The primary aim of this study was to use proteomic analysis to identify plasma proteins with altered abundance among T2DM patients following the onset of AMI compared with patients without AMI.
Materials and methods
Recruitment of subjects
This study was conducted among diabetic Malaysian subjects according to the Declaration of Helsinki and approved by the Ethical Committee (Institutional Review Board) of the University of Malaya Medical Centre (UMMC) (REF NO 1003.10(1)) and Faculty of Dentistry, University of Malaya (DF OB1404/0020(L)). Experimental samples used in the study were collected from patients admitted to the UMMC Cardiology and Coronary Care Wards, and control samples were collected from patients attending the outpatients' clinic at UMMC Medical Clinic. Samples from admitted patients were collected within three days following onset of AMI, depending on the patients' medical status. Each volunteer was interviewed and requested to complete a questionnaire concerning personal and health information, including smoking and alcohol drinking habits, health and dental problems, radiation exposure, major surgical procedures, and antibiotic prescriptions. Informed written consent was obtained from every patient prior to plasma collection. Blood samples were obtained by peripheral venous puncture and collected into plastic whole blood tubes with spray-coated dipotassium EDTA. Within 1 h of collection, the blood samples were centrifuged at 1500×g for 15 min at 4 °C. The resulting supernatant, or plasma, was immediately divided into 100 μL aliquots and stored at -80 °C. Following 15 months of subject enrollment, 43 admitted patients and 34 outpatients were recruited for this study. The exclusion criteria for recruited subjects were pregnancy, smoking during the previous three months, antibiotic use during the previous three months, and prior major surgeries. In accordance with these exclusion criteria, plasma samples from 20 subjects were finally chosen for proteomic analysis. The demographics and clinical characteristics of subjects involved in the study are shown in Table 1.
Separation of proteins by two-dimensional gel electrophoresis
Initially, 4.5 μL (approximately 315 μg of protein) of plasma was incubated in sample buffer (9 mol/L urea, 60 mmol/L DTT, 2% v/v IPG buffer pH 4–7, 0.5% v/v Triton X-100) for 30 min and then in rehydration buffer (8 mol/L urea, 0.5% v/v IPG buffer pH 4–7, 0.5% v/v Triton X-100, 0.002% w/v Orange G) for another 30 min at room temperature, with a final volume of 200 μL. The final ratio of sample buffer to rehydration buffer was 1:3. Once the mixture was completely dissolved, one Immobiline DryStrip Gel (pH 4–7 linear, 11 cm, GE Healthcare, Uppsala, Sweden) was passively rehydrated with the mixture and kept in a sealed environment for a minimum of 12 h at room temperature. Isoelectric focusing (IEF) was performed on the other strips with an Ettan IPGphor 3 Isoelectric Focusing Unit (GE Healthcare, Uppsala, Sweden), and they were maintained at 20 °C. With a maximum current of 50 μA per strip, the rehydrated IPG gel strips were electrophoresed at 500 V for 1 h (step and hold), 1000 V for 1 h (gradient), 6000 V for 2.5 h (gradient), and 6000 V for 55 min (step and hold).
Prior to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), one 8%–18% gradient SDS-PAGE gel with a total volume of approximately 24 mL was prepared for each strip. Upon completion of IEF, the focused strips were equilibrated for 15 min in SDS equilibration buffer (75 mmol/L Tris, pH 8.8, 6 mol/L urea, 29.3% v/v glycerol, 2% w/v SDS) containing 1% w/v DDT on an orbital shaker. The solution was then replaced by equilibration buffer containing 4.5% w/v iodoacetamide for another 15 min with gentle shaking. Subsequently, SDS-PAGE was performed using an SE 600 Ruby Electrophoresis System (GE Healthcare, Uppsala, Sweden) filled with anode buffer, pH 8.8 (45.4 mol/L Tris), at 18 °C on 8%-18% gradient gels containing SDS with the strips sealed in place with 0.5% w/v agarose in SDS-electrophoresis buffer (25 mmol/L Tris, 192 mmol/L glycine, 0.1% w/v SDS). SDS-PAGE was performed at 50 V for 0.5 h, followed by 600 V for 1.5 h. The maximum current and power were 40 mA and 25 W per gel, respectively.
An MS-compatible silver staining protocol was performed according to the method previously described, with slight modifications13. The procedure was carried out on an orbital shaker, as all of the steps in the staining technique required gentle shaking. After gel electrophoresis, the gel slabs were soaked in fixing solution (40% v/v ethanol, 10% v/v acetic acid) for at least 30 min. The gels were then incubated in sensitizing solution (30% v/v ethanol, 0.5 mol/L sodium acetate trihydrate, 8 mmol/L sodium thiosulfate) for at least another 30 min. Following three consecutive 15 min washes with double-distilled water, the solution was substituted with silver solution (14.72 mmol/L silver nitrate) and left to shake for 20 min. Subsequently, the gel slabs were quickly washed twice and immersed in developing solution (235.9 mmol/L sodium carbonate, 0.2% v/v formaldehyde). The protein spots were developed until a sufficient resolution was reached. The solution was then replaced with stop solution (39.2 mmol/L EDTA disodium dihydrate) to prevent overstaining. After washing with double-distilled water three times, the developed gel slabs were then stored at 4 °C.
Analysis of gel images
After staining, the 2DE gels were scanned at 600 dpi using ImageScanner III (GE Healthcare, Uppsala, Sweden). Image analysis was performed using Progenesis SameSpots (version 4.5, Nonlinear Dynamics, UK & USA). Ten gel images with the best resolution were chosen to represent each group and used for further analysis. In brief, the gel images were aligned to adjust all of the spots accurately in the exact same location. After calculating the background-corrected abundance, the normalized volume was calculated for each protein spot that was detected by the software. The volumes for both groups were compared to determine the respective fold changes and P-values (unpaired t-test) of the protein spots as calculated by the image analysis software. Each protein spot of altered abundance was expressed as averaged normalized volume±standard error of the mean (SEM). The P-values later underwent false discovery rate (FDR) analysis based on the method described by Benjamini-Hochberg14. This statistical analysis was performed with the help of a statistician, and the formula was prepared in a Microsoft Excel file at an alpha value of 0.05. Each of the highly resolved plasma protein spots that was statistically significant (adjusted P≤0.050) with fold changes of at least 1.5 was subsequently selected for MS analysis. As a result, 34 plasma spots were selected for subsequent experiments. The protein spots, ranging from 1 to 2 mm in diameter with the desired significance level were manually excised from the silver-stained 2DE gels and pooled from all similar gels to maximize the confident MS results. The gel plugs were then kept hydrated in double-distilled water for further protein identification.
Identification of proteins by MALDI TOF/TOF
For in-gel tryptic digestion, the gel plugs were destained with 15 mmol/L potassium ferricyanide/50 mmol/L sodium thiosulfate for 15 min with shaking until they turned transparent. The gel plugs were then reduced in 10 mmol/L DTT/100 mmol/L ammonium bicarbonate for 30 min at 60 °C, followed by alkylation in 55 mmol/L iodoacetamide/100 mmol/L ammonium bicarbonate for 20 min in the dark at room temperature. The plugs were then washed three times with 50% acetonitrile/100 mmol/L ammonium bicarbonate for 20 min with shaking, followed by dehydration with 100% acetonitrile for 15 min with shaking and centrifugation in a speed vacuum at low speed at ambient temperature until the gel plugs were completely dry. The plugs were digested in 6 ng/μL trypsin in 50 mmol/L ammonium bicarbonate at 37 °C. On the following day, the peptide mixtures were extracted twice with 50% and 100% acetonitrile sequentially and concentrated in a speed vacuum. The dried peptides were subsequently reconstituted with 0.1% formic acid and desalted using Zip Tip C18 Micropipette Tips.
Peptide mixtures were analyzed using a 5800 MALDI TOF/TOF Analyzer (AB SCIEX, USA). The tryptic-digested peptides were crystallized with an α-cyano-4-hydroxycinnamic acid matrix solution (6 mg/mL α-cyano-4-hydroxycinnamic acid, 70% acetonitrile, 0.1% v/v TFA aqueous solution) and spotted onto a 384-well MALDI target plate. The MS results were automatically acquired with a trypsin autodigest exclusion list, and the 20 most intense precursor ions were selected for MS/MS analysis, with a minimum S/N of at least 10. MS and MS/MS acquisition and interpretation were carried out using TOF/TOF Series Explorer Software (version 4.0, AB SCIEX, USA). The spectra were then processed and analyzed using ProteinPilot Software (version 4.5, AB SCIEX, USA) and the in-house MASCOT Program (Matrix Science, UK) to search for peptide mass fingerprints and MS/MS data. Both combined MS and MS/MS searches were conducted against the UniProt database (Last updated: July 31, 2017) with the following search parameters: Homo sapiens; trypsin enzyme; one missed cleavage; peptide mass tolerance at 100 ppm; fragment mass tolerance at 0.2 Da; fixed and variable modifications, including cysteine carbamidomethylation and methionine oxidation, respectively; and inclusion of monoisotopic masses. According to the MASCOT search results, protein scores greater than 54 were considered significant (P<0.05).
Validation of protein concentration by ELISA
Analysis of the identified proteins and respective cardiac associations from a text-mining database15 led to the further selection of plasma tetranectin and titin for analysis by competitive ELISA. This immunoassay employed the quantitative sandwich enzyme immunoassay technique. Based on the manufacturer's instructions, protein determination was performed on two antibodies specific to tetranectin and titin. In principle, the microtiter plate provided by the manufacturer was pre-coated beforehand with a specific antibody. For each protein, ten samples and the standard were pipetted into the appropriate microtiter plate wells. In theory, any desired analyte present in the samples was bound by the immobilized antibody. Upon removing any unbound substances, a biotin-conjugated antibody specific to its analyte was added. After washing, avidin conjugated to horseradish peroxidase was added to each microplate well and incubated. Following a wash to remove any unbound avidin-enzyme reagent, 3, 3', 5, 5'-tetramethylbenzidine substrate was added to the wells. Only those wells containing the desired analyte, biotin-conjugated antibody, and enzyme-conjugated avidin exhibited a change in color. The intensity of the developed color was proportional to the amount of analyte bound in the initial step. The color development of the enzyme-substrate reaction was terminated by the addition of sulfuric acid solution, and the color intensity was measured spectrophotometrically at a wavelength of 450 nm. The concentration of analyte in the samples was then determined by comparing the optical density of the samples to the standard curve generated for each antigen.
Results
Analysis of plasma proteins
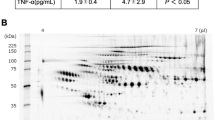
Profiling of human plasma samples by 2DE separated hundreds of protein spots based on different isoelectric points and relative molecular masses. A typical representative 2DE profile of plasma within the range of pH 4–7 with the relative molecular mass declining from 200 kDa to 10 kDa was obtained, as demonstrated in Figure 1A. When 2DE was performed on plasma samples from diabetic patients with AMI, the profiles obtained were similar to those of the controls. The 2DE plasma protein profiles resolved in the present study were also generally comparable with those published in the Swiss-2D PAGE database16. For enhanced visualization and comparison, individual protein spots between the two groups were enlarged, as shown in Figure 1B. The spots were obtained from the software and tabulated according to the respective protein labels. By using Progenesis SameSpots and FDR analysis, a total of 34 highly resolved protein spots were differentially regulated (adjusted P≤0.050). Densitometry analysis of the highly resolved spots was conducted in terms of the average normalized volumes, P-values, and fold changes. Protein spots that were statistically significant (adjusted P≤0.050) with a minimum fold changes of 1.5 upon FDR analysis are recorded in Table 2. Of these protein spots, 10 were significantly upregulated and 24 were significantly downregulated in T2DM patients with AMI compared with T2DM patients without AMI. These 34 protein spots of altered abundance comprised a total of 17 plasma protein clusters.

Representative 2DE profile of the plasma proteome and cropped images of protein spots of altered abundance. Ten gels of each group of T2DM patients were analyzed by Progenesis SameSpots, and one of the gels was selected as a representative (A). Thirty-four spots of altered abundance were labelled and circled in red: 10 were significantly upregulated (Protein spots 6, 9, 10, 11, 12, 13, 14, 24, 27, and 31) and 24 were significantly downregulated (Protein spots 1, 2, 3, 4, 5, 7, 8, 15, 16, 17, 18, 19, 20, 21, 22, 23, 25, 26, 28, 29, 30, 32, 33, and 34). Representative cropped images of T2DM patients with (+AMI) and without (−AMI) AMI are shown in (B).
Identification of plasma proteins of altered abundance
The identities of the protein spot clusters were confirmed by MS and a database search. Every spot with altered abundance in the 2DE gel slabs was subjected to MS analysis for identification. According to the MASCOT search results, protein scores greater than 54 were considered significant (P<0.05). The results for plasma proteins were enumerated in terms of their UniProtKB accession number, theoretical pI, theoretical mass, MASCOT score, number of peptides matched, and sequence coverage (Table 3). MS was able to identify 12 proteins with notable changes between the experimental and control groups, including clusterin (4 protein species), haptoglobin (6 protein species), DNA repair protein RAD50 (1 protein species), serum albumin (4 protein species), apolipoprotein A-IV (2 protein species), serotransferrin (1 protein species), tetranectin (1 protein species), titin (1 protein species), retinol-binding protein 4 (2 protein species), transthyretin (1 protein species), alpha-1-antitrypsin (1 protein species), and apolipoprotein A-I (2 protein species). Eight protein spots, however, were not statistically significant: clusterin, myosin-13, zinc finger protein 445, aminoacyl tRNA synthase complex-interacting multifunctional protein 1, caspase-6 and serum amyloid A, and this is most likely due to the limited gel plugs and low protein concentration. With the exceptions of haptoglobin and myosin-13, the theoretical pI and mass values of most of the protein spots of interest were apparently comparable with the values detected from their resolved positions in the 2DE profiles shown in Figure 1. Variations in the haptoglobin and myosin-13 values may be due to post-translational modifications and/or fragmentation of the proteins.
Validation of the plasma proteins of interest using ELISA
When the plasma proteins of altered abundance were subjected to text mining using the method of Pletscher-Frankild et al 16, relevant clinical associations were generated (Table 4). According to the literature, most of the identified plasma proteins of interest had already been associated with cardiac coronary artery diseases (CADs) or atherosclerosis. From this list, tetranectin and titin were further selected for quantitative validation by sandwich ELISA. While tetranectin was chosen mainly because of its association with fibrinolysis, the selection of titin was based on its relationship with cardiac and muscle diseases and because it is a resident cardiac protein. The regulation patterns and significance levels of tetranectin and titin obtained by ELISA and 2DE were compared (Figure 2). ELISA was performed on ten samples from each group, and these proteins were confirmed to be present at different concentrations. From these concentrations, the abundance patterns between both the control (without AMI) and experimental groups (with AMI) were calculated and found to be similar to the initial 2DE data. Ultimately, the concentrations of the two proteins were confirmed through this method, and the ELISA and 2DE approaches both demonstrated downregulation of tetranectin and upregulation of titin in diabetic subjects with AMI.

Analysis of plasma tetranectin and titin by 2DE and ELISA. Similar patterns of plasma tetranectin and titin were obtained using 2DE (A) and ELISA (B). However, the P-value for tetranectin was not as significant as that obtained through the 2DE approach. : T2DM patients without AMI, : T2DM patients with AMI.
Discussion
Most of the identified proteins of altered abundance were linked with broad disease associations resulting from the inflammatory response, blood coagulation, acute-phase response, cholesterol homeostasis, cardiac muscle tissue development, and other pathophysiological responses (Universal Protein Resource, 2017; http://www.uniprot.org/). Some, including tetranectin and titin, were reportedly associated with cardiovascular disease and diabetes. Due to their associations with cardiac metabolism pathways and presence in cardiac muscles, tetranectin and titin were further selected for concentration validation using ELISA. The results clearly showed regulation patterns similar to those found by the 2DE analysis, with slightly different fold changes. The significance level for tetranectin was, however, much lower than that calculated from the 2DE analysis. The different P values may be attributable to the presence of isoforms and/or PTMs of these proteins or the specificity of the antibodies used for ELISA. The significance level may be improved by increasing the number of samples and/or using antibodies that are highly specific to fragments and isoforms from both the control and experimental groups.
Tetranectin is a C-type lectin and an endogenous ligand that specifically binds to the plasminogen kringle 4 domain, thereby enhancing plasminogen activation17. It is primarily involved in tissue remodeling and development18. Based on the generated 2DE profile, the tetranectin detected was close to its theoretical mass (23 kDa). It is possible that the downregulation of tetranectin in plasma is due to its fibrinolytic property in thrombus breakdown. Hence, tetranectin dissolves the thrombus blocking the coronary artery and promotes better blood circulation to cardiac muscles. This implied role of tetranectin in the fibrinolytic process suggests that it has a functional role in AMI patients. Further evaluation and correlative studies of cardiac tetranectin in the formation and dissolution of fibrin clots are highly recommended.
Tetranectin, as a new regulator of fibrinolysis, has been studied by Kamper et al using plasma from 60 AMI patients and 30 healthy subjects to investigate its possible involvement in the pathophysiology of AMI19. This population study provides clear evidence of a significant decrease of tetranectin levels in AMI patients compared with healthy subjects. Likewise, another MS study performed by Yin et al showed comparable results; the tetranectin levels were inversely correlated with the risk of atherosclerotic cardiovascular disease20. More recently, Chen et al also confirmed that the serum tetranectin levels in patients suffering from stable CAD were decreased compared with those of healthy controls21. These studies are in agreement with the results obtained in this research.
Titin is a large protein that is longer than 1 μm in length and the third most abundant muscle protein after myosin and actin. Also known as connectin, titin functions as a molecular spring and is responsible for the passive elasticity of muscle22. In fact, titin is a muscle-specific protein that is found in cardiac and skeletal muscles. It is essential for the contraction of striated muscle tissues23. Titin plays a crucial role in the temporal and spatial control of the assembly and elasticity of the highly ordered sarcomeres of the striated muscle24. Titin limits the range of motion of the sarcomere in tension, thus contributing to the passive stiffness of muscle. By providing connections at the level of individual microfilaments, titin contributes to the careful balance of forces between the halves of a sarcomere and ultimately maintains sarcomere integrity. Variations in the sequence of titin in different types of muscles, including cardiac and skeletal muscles, have been correlated with differences in the mechanical properties of these muscles. According to the UniProt database (Universal Protein Resource, 2017), titin is robustly linked with diverse cardiac processes, including blood coagulation, cardiac muscle contraction, cardiac muscle fiber development, cardiac muscle hypertrophy, cardiac muscle tissue morphogenesis, cardiac myofibril assembly, detection of muscle stretch, and many others.
The about 35 kDa titin spot that was detected in the 2DE profile is most likely a truncated fragment of the native 3843 kDa molecule. It is possible that the upregulation of titin in plasma is due to its dissociation from cardiac muscle when myocardial infarction is caused by prolonged ischemia. When tissue death occurs, the contraction of the striated cardiac muscle tissue is altered. This pattern is in keeping with the clinical evaluation performed by Vassiliadis et al 25. In their study, the investigators found that a statistically significant increase in the level of a titin fragment (NVTVEARLIK) was due to the degradation of titin by MMP-12 in AMI subjects. Titin is the first neoepitope biomarker that was specifically detected in serum and related to cardiac damage. In another serological study, the presence of the N2B exon, a cardiac-specific titin fragment, in blood has been suggested to be a sensitive marker for myocardial injury26. Additionally, changes in titin phosphorylation and titin splicing occur in cardiac disease despite mutations in the titin gene27, demanding further study of this protein to elucidate the specific machinery responsible for titin modifications.
The preliminary findings in this study have potential in clinical applications. The about 35 kDa plasma titin fragment may be an additional diagnostic biomarker for AMI. Recently, troponin has become central to the definition of AMI in the consensus guidelines from the European Society of Cardiology and the American College of Cardiology (Medscape, 2017; http://emedicine.medscape.com/article/811905-overview). In general, a cardiac troponin (cTn) test is the most common test in a clinical setting, compared with tests of creatine kinase, lactate dehydrogenase, aspartate transaminase, myoglobin, ischemia modified albumin, pro-brain natriuretic peptide, and glycogen phosphorylase isoenzyme BB. Over time, identification of more proteins can lead to insight into the usefulness of combinatorial biomarkers associated with AMI. For instance, copeptin not only identifies patients at risk of mortality but its addition to cTn also improves sensitivity and rules out AMI. Thus, copeptin may help identify patients who may be safely discharged early from the emergency department28.
Apart from finding valuable diagnostic biomarkers, this proteomic study aimed to identify possible biomarkers prior to the onset of AMI. While the biological reasons for the changes in protein levels are not quite understood, AMI-linked tetranectin and titin have the potential to be used in early screening for heart disease. In the future, a longitudinal cohort study should be performed on similar patients at high risk for AMI to investigate whether tetranectin and titin can be used as biomarkers to predict cardiac arrest. The tetranectin level may indicate the status of cardiac thrombosis, whereas the titin level may reflect the severity of cardiac tissue death in people with AMI high-risk factors. Early screening, particularly among high-risk patients, can be made possible with a test kit designed to detect these proteins in a specified range of concentrations. The test kit will be suitable for use in diabetic clinics and hospitals if the proteins are found to be associated with T2DM groups with susceptibility to AMI.
In conclusion, tetranectin and titin in plasma may be useful as biomarkers for the prediction of AMI, particularly in high-risk T2DM patients.
Authors contribution
Mohd Aizat Abdul RAHIM performed the study, analyzed the data, and wrote the manuscript; Zubaidah Haji Abdul RAHIM designed the study and revised the manuscript; Wan Azman WAN AHMAD, Marina Mohd BAKRI, Muhammad Dzafir ISMAIL provided clinical samples and revised the manuscript from a clinical perspective; Onn Haji HASHIM designed the study, contributed analytic tools, and critically revised the manuscript. All of the authors approved the final manuscript.
References
Mendis S, Armstrong T, Bettcher D, Branca F, Lauer J, Mace C, et al. Executive summary. In: World Health Organization, editor. Global status report on noncommunicable diseases 2014. Switzerland: World Health Organization; 2014. p 9–32.
Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339: 229–34.
Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract 2014; 103: 137–49.
Moon A, Bhale N, Shinde GB, Sahare P, Talreja K. Protein biomarkers: in life and after life. J Adv Med Life Sci 2014; 1: 1–6.
Ceciliani F, Eckersall D, Burchmore R, Lecchi C. Proteomics in veterinary medicine: applications and trends in disease pathogenesis and diagnostics. Vet Pathol 2014; 51: 351–62.
Legrain P, Aebersold R, Archakov A, Bairoch A, Bala K, Beretta L, et al. The human proteome project: current state and future direction. Mol Cell Proteomics 2011; 10: 1–5.
Gorg A, Weiss W, Dunn MJ. Current two-dimensional electrophoresis technology for proteomics. Proteomics 2004; 4: 3665–85.
Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature 2003; 422: 198–207.
Elek G, Lapis K. A path or a new road in laboratory diagnostics? Biological mass spectrometry: facts and perspectives. Pathol Oncol Res 2006; 12: 179–83.
Kristensen LP, Larsen MR, Mickley H, Saaby L, Diederichsen AC, Lambrechtsen J, et al. Plasma proteome profiling of atherosclerotic disease manifestations reveals elevated levels of the cytoskeletal protein vinculin. J Proteomics 2014; 101: 141–53.
Thambisetty M, Lovestone S. Blood-based biomarkers of Alzheimer's disease: challenging but feasible. Biomark Med 2010; 4: 65–79.
Kossaify A, Garcia A, Succar S, Ibrahim A, Moussallem N, Kossaify M, et al. Perspectives on the value of biomarkers in acute cardiac care and implications for strategic management. Biomark Insights 2013; 8: 115–26.
Yan JX, Wait R, Berkelman T, Harry RA, Westbrook JA, Wheeler CH, et al. A modified silver staining protocol for visualization of proteins compatible with matrix-assisted laser desorption/ionization and electrospray ionization-mass spectrometry. Electrophoresis 2000; 21: 3666–72.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc 1995; 57: 289–300.
Pletscher-Frankild S, Pallejà A, Tsafou K, Binder JX, Jensen LJ. DISEASES: Text mining and data integration of disease–gene associations. Methods 2015; 74: 83–9.
Sanchez JC, Appel RD, Golaz O, Pasquali C, Ravier F, Bairoch A, et al. Inside SWISS-2DPAGE database. Electrophoresis 1995; 16: 1131–51.
Mogues T, Etzerodt M, Hall C, Engelich G, Graversen JH, Hartshorn KL. Tetranectin binds to the kringle 1-4 form of angiostatin and modifies its functional activity. J Biomed Biotechnol 2004; 2004: 73–8.
Jaquinod M, Holtet TL, Etzerodt M, Clemmensen I, Thogersen HC, Roepstorff P. Mass spectrometric characterisation of post-translational modification and genetic variation in human tetranectin. Biol Chem 1999; 380: 1307–14.
Kamper EF, Kopeikina L, Mantas A, Stefanadis C, Toutouzas P, Stavridis J. Tetranectin levels in patients with acute myocardial infarction and their alterations during thrombolytic treatment. Ann Clin Biochem 1998; 35: 400–7.
Yin X, Subramanian S, Hwang SJ, O'Donnell CJ, Fox CS, Courchesne P, et al. Protein biomarkers of new-onset cardiovascular disease: prospective study from the systems approach to biomarker research in cardiovascular disease initiative. Arterioscler Thromb Vasc Biol 2014; 34: 939–45.
Chen Y, Han H, Yan X, Ding F, Su X, Wang H, et al. Tetranectin as a potential biomarker for stable coronary artery disease. Sci Rep 2015; 5: 1–8.
Labeit S, Gautel M, Lakey A, Trinick J. Towards a molecular understanding of titin. EMBO J 1992; 11: 1711–6.
Tskhovrebova L, Trinick J. Roles of titin in the structure and elasticity of the sarcomere. J Biomed Biotechnol 2010; 2010: 1–7.
Mayans O, van der Ven PF, Wilm M, Mues A, Young P, Furst DO, et al. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature 1998; 395: 863–9.
Vassiliadis E, Rasmussen LM, Byrjalsen I, Larsen DV, Chaturvedi R, Hosbond S, et al. Clinical evaluation of a matrix metalloproteinase-12 cleaved fragment of titin as a cardiovascular serological biomarker. J Transl Med 2012; 10: 1–10.
Bogomolovas J, Gasch A, Bajoras V, Karčiauskaitė D, Šerpytis P, Grabauskienė V, et al. Cardiac specific titin N2B exon is a novel sensitive serological marker for cardiac injury. Int J Cardiol 2016; 212: 232–4.
LeWinter MM, Granzier H. Cardiac titin: a multifunctional giant. Circulation 2010; 121: 2137–45.
Lipinski MJ, Escarcega RO, D'Ascenzo F, Magalhaes MA, Baker NC, Torguson R, et al. A systematic review and collaborative meta-analysis to determine the incremental value of copeptin for rapid rule-out of acute myocardial infarction. Am J Cardiol 2014; 113: 1581–91.
Acknowledgements
This study was supported by the PG110-2014A and HIR-MOHE H-20001-00-E000009 research grants from the University of Malaya, and Young Lecturer's Scheme awarded to MAAR by Universiti Teknologi MARA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Rahim, M., Rahim, Z., Ahmad, W. et al. Inverse changes in plasma tetranectin and titin levels in patients with type 2 diabetes mellitus: a potential predictor of acute myocardial infarction?. Acta Pharmacol Sin 39, 1197–1207 (2018). https://doi.org/10.1038/aps.2017.141
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.141
Keywords
This article is cited by
-
Tetranectin as a potential novel prognostic biomarker in anthracycline-related cardiac dysfunction
Heart and Vessels (2023)
-
Tetranectin, a potential novel diagnostic biomarker of heart failure, is expressed within the myocardium and associates with cardiac fibrosis
Scientific Reports (2020)
-
Circulating biomarkers for cardiovascular diseases: the beats never stop
Acta Pharmacologica Sinica (2018)
