
Abstract
Aim:
JG6 is a novel marine-derived oligosaccharide that has shown to inhibit angiogenesis and tumor metastasis. In this study, we sought to identify the potential target responsible for the anti-cancer activity of JG6.
Methods:
Human liver cancer cell line Bel-7402 and human cervical cancer cell line HeLa were examined. CXCL12-stimulated cell proliferation and migration were determined using a CCK-8 kit and a transwell assay, respectively. Western blotting was performed to examine the changes in CXCL12/CXCR4 axis. Molecular docking and surface plasmon resonance (SPR) were performed to characterize the possible interaction between JG6 and the CXCL12/CXCR4 axis.
Results:
Treatment with CXCL12 potently stimulated the proliferation and migration in both Bel-7402 and HeLa cells. Co-treatment of the cells with JG6 (10, 50 and 100 μg/mL) dose-dependently impeded the CXCL12-stimulated cell proliferation and migration. Furthermore, CXCL12 rapidly induced phosphorylation of AKT, ERK, FAK and Paxillin in Bel-7402 and HeLa cells, whereas pretreatment with JG6 dose-dependently inhibited the CXCL12-induced phosphorylation of these proteins. The SPR assay showed that JG6 bound to CXCL12 with a high affinity. In molecular docking study, JG6 appeared to interact with CXCL12 via multiple polar interactions, including 6 ionic bonds and 7 hydrogen bonds.
Conclusion:
Inhibition of the CXCL12/CXCR4 axis by JG6 may account for its anticancer activity.
Similar content being viewed by others


Introduction
CXCL12, also called stromal cell-derived factor-1 (SDF-1), is a CXC chemokine that binds primarily to CXC receptor 4 (CXCR4). This binding induces intracellular signaling that plays a pivotal role in chemotaxis, cell survival, proliferation, and metastasis of CXCR4+ tumor cells to organs that express CXCL121,2. It has been reported that CXCR4+ cancers metastasize to bones and lymph nodes in a CXCL12-dependent manner3,4. Therefore, the CXCL12/CXCR4 axis has emerged as a potential target for cancer therapy.
Increasing effort has been made in impeding the function of the CXCL12/CXCR4 axis by targeting either the ligand or the receptor using small-molecule receptor antagonists, receptor-binding antibodies, or modified chemokines5,6. Among these options, blocking the access of CXCL12 to CXCR4 is an effective approach. It has been reported that glycosaminoglycans (GAGs) and syndecan-4 hindered the invasion of human tumor cells (HeLa cells) via blocking the interaction between CXCL12 and CXCR47, and this finding was supported by the structural analysis of CXCL12's interactions with GAGs8,9. Accordingly, heparin, a sulfated glycosaminoglycan, blocked CXCL12-induced human cancer cell growth, migration, and invasion7,10. Furthermore, treatment of human hepatoma cells with a series of GAG mimetics inhibited CXCL12-induced migration and invasion11.
JG6 is a novel marine-derived oligosaccharide discovered in our laboratory. JG6 significantly suppressed cancer growth and metastasis in vitro and in vivo12,13, but the mechanisms remain unclear. Importantly, we have noted that the structure of JG6 resembles the structure of GAGs. Here, we explored whether JG6 has potential anti-cancer activity through its intervention in the CXCL12/CXCR4 axis.
Materials and methods
Reagents
JG6 was created by semi-synthesis followed by sulfate modification by reacting oligomannurarate from sodium alginate with ClSO3H in formamide. The pH of the products was adjusted to 7.0 with 4 mol/L NaOH and desalted using Sephadex G-10. The products were pooled and freeze-dried. Analysis was performed using High Performance Gel Permeation Chromatography (HPGPC) with a G3000W×1 column (300 mm×7.8 mm) (TOSOH, Japan). The structure is depicted in Figure 1. JG6 is composed of β-(1–4) glycosidic linked D-mannurarate with sulfate modification of the hydroxyl group at the C2/C3 position. CXCL12 was purchased from R&D Systems (Minneapolis, MN, USA).

Chemical structure of JG6.
Cell culture
Bel-7402 human liver cancer cells and HeLa human cervical cancer cells were obtained from the Institute of Biochemistry and Cell Biology and the American Type Culture Collection, respectively. The cells were grown in RPMI-1640 or high-glucose DMEM medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin). The cells were maintained in a 5% CO2 humidified atmosphere at 37 °C.
Proliferation assay
Bel-7402 and HeLa cells were incubated in serum-free medium for 6 h and then exposed to various concentrations of JG6 (0, 10, 50, or 100 μg/mL) and CXCL12 (300 ng/mL) for 72 h. Cell proliferation was determined using a CCK-8 kit (Dojindo, Kumamoto, Japan).
Migration assay
The cell migration assay was performed in a transwell Boyden Chamber (8 μm pore size, Costar, MA, USA). Briefly, tumor cells (3.5×105 cells per well) were seeded in the top chamber of the transwell, whereas the lower chamber was filled with culture medium supplemented with 50 or 300 ng/mL CXCL12 plus JG6 (0, 10, 50, or 100 μg/mL). CXCL12 was not added to the negative control group. Cells were cultured for 24–36 h at 37 °C in a well-humidified incubator, and then the total migrated cells were fixed, stained with 0.1% crystal violet, and imaged under a microscope. Stained cells were extracted with 10% acetic acid and the OD value was measured at 595 nm.
Western blotting analysis
Cells were starved for 24 h, pre-incubated with JG6 (0, 10, 50, or 100 μg/mL) for 6 h, and then induced with CXCL12 (R&D, Minneapolis, MN, USA) at 50 ng/mL for 15 min. After incubation, cells were lysed in 1× SDS lysis buffer (50 mmol/L Tris-HCl pH 6.8, 100 mmol/L DTT, 2% SDS, 0.1% bromophenol blue, and 10% glycerol), and then the lysates were clarified by centrifugation at 14 000×g for 10 min. Equal amounts of protein were subjected to SDS-PAGE and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, USA). The membranes were blocked with 5% nonfat milk and incubated with the corresponding primary antibodies at 4 °C overnight. The immunoreactive bands were visualized by the SuperSignal West PicoChemiluminescent kit (Pierce Biotechnology, Rockford, IL, USA) using Image Quant LAS4000 (GE, USA). Primary antibodies used include antibodies specific for Phospho-Paxillin (Y118), Paxillin, Fak, phospho-Erk1/2, Erk1/2, phospho-Akt (Ser473) and Akt (Cell Signaling Technology, USA), p-Fak (pY861) (Invitrogen, USA), CXCL12 and CXCR4 (Abcam, UK).
Molecular docking
The NMR structure of chemokine CXCL12 was retrieved from the Brookhaven Protein Data Bank (PDB entry: 2K03). Chain A and chain C from 2K03 were retained as the receptor, whereas chain B was selected to define the grid box center. Molecular docking was performed using Glide with the XP mode14,15. The compounds were sketched by Maestro and then processed by LigPrep with Epik to treat the protonation states. The docked conformations in the ligand-binding site were selected for further analysis.
Surface plasmon resonance analysis
The kinetics and specificity of the binding reaction between JG6 and CXCL12 were analyzed using the BIAcore surface plasmon resonance apparatus. Briefly, JG6 was immobilized on an NLC sensor chip according to the Ligand Thiol protocol described in BIA application handbook. To assess real-time binding capacity, 5–80 nmol/L of CXCL12 was injected over the sensor chip surface with the immobilized JG6 and then washed with HBS-EP buffer for 5 min. The sensor chip surface was regenerated using 60 μL of NaCl (2 mol/L). All binding experiments were performed at 25 °C with a constant flow rate of 10 μL/min of HBS-EP. For binding affinity assessment, the association phase was allowed to proceed to equilibrium.
The untreated surface moieties were blocked with ethanolamine. To correct for nonspecific binding and bulk refractive index changes, a blank channel (FC2) without oligosaccharide was employed as a control for each experiment. Sensorgrams for all binding interactions were recorded in real time, and the blank channel readings were subtracted from the results before analysis. Changes in mass due to the binding response were recorded as resonance units (Ru). Binding kinetics and affinities were determined by SPR using BIAcore software 3.1.
Statistical analysis
The data are presented as the mean±SD. Statistical significance was analyzed by two-tailed Student's t-test. A value of P<0.05 was considered significant.
Results
JG6 impeded CXCL12-induced cancer cell proliferation
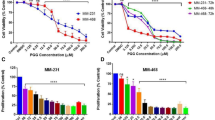
The CXCL12/CXCR4 biological axis plays an important role in regulating tumor cell proliferation. First, we investigated whether JG6 had an impact on CXCL12-stimulated cell growth. The CCK-8 assay was applied to measure the cell proliferation rates of Bel-7402 cells and HeLa cells. CXCL12 stimulated tumor cell growth, as previously reported10. The proliferation rate was increased by almost 50% upon treatment with 300 ng/mL CXCL12 (Figure 2A). This effect was largely reversed when cells were simultaneously treated with JG6. JG6 caused a dose-dependent suppression of CXCL12-stimulated Bel-7402 cell proliferation. The cell proliferation rate was inhibited by 22.2% at 10 μg/mL, 46.5% at 50 μg/mL, and 51.0% at 100 μg/mL. Similar results were observed in HeLa cells, for which treatment with 10, 50, or 100 μg/mL JG6 yielded 22.2%, 37.2%, and 41.5% lower cell proliferation rates, respectively, than in the absence of JG6 (Figure 2A). However, JG6 did not inhibit Bel-7402 and HeLa cell proliferation in vitro in normal cell culture conditions (data not shown).

JG6 blocked CXCL12-induced cancer cell proliferation and migration in vitro. (A) JG6 inhibited cancer cell proliferation induced by CXCL12. (a) CXCL12 induced Bel-7402 (left) and HeLa (right) cell proliferation. Bel-7402 (b) and HeLa (c) cells were treated with CXCL12 (300 ng/mL) and 0, 10, 50, or 100 μg/mL JG6 for 72 h. (B, C) JG6 suppressed the cell migration induced by CXCL12 in Bel-7402 (B) and HeLa cells (C). Representative image of cell migration were displayed (magnificaction: 200×; +: treatment with CXCL12). The rate of cell migration inhibition by JG6 were shown as percentage of CXCL12 treatment (mean±SD, from three independent experiments. bP<0.05, cP<0.01 vs control).
JG6 suppressed CXCL12-induced cancer cell migration
The CXCL12/CXCR4 biological axis is also known to modulate the chemotactic motility of cancer cells. We next evaluated whether JG6 had an inhibitory effect on this process. As shown in Figure 2B, Bel-7402 cells were responsive to CXCL12 induction at a JG6 concentration of 50 ng/mL for 48 h when a notable increase in migrating cells was observed compared to the negative control group (which was incubated without CXC12 and FBS). JG6 inhibited CXCL12-induced migration of Bel-7402 cells in a dose-dependent manner, yielding 100% inhibition at 100 μg/mL. Likewise, although HeLa cells were less responsive to CXCL12-stimulation than Bel-7402 cells, JG6 similarly reduced CXCL12-induced migration in HeLa cells by 44%, 88%, and 97% after treatment with 10, 50, or 100 μg/mL of JG6 (Figure 2C). However, JG6 did not impede Bel-7402 cell migration induced by 10% FBS (data not shown).
JG6 blocked CXCL12-induced signal transduction
We showed that JG6 effectively inhibited CXCL12-induced cell proliferation and migration in Bel-7402 and HeLa cells. We then proceeded to explore its effect on downstream signals of the CXCL12/CXCR4 axis. The Western blotting data presented in Figure 3 show that 50 ng/mL CXCL12 rapidly induced phosphorylation of AKT, ERK, FAK and Paxillin in Bel-7402 and HeLa cells, within 15 min. In contrast, pre-incubation with JG6 for 6 h inhibited CXCL12-induced phosphorylation of these proteins in a dose-dependent manner. Similar results were observed in HeLa cells.

JG6 blocked CXCL12/CXCR4 axis signal transduction in Bel-7402 (A) and HeLa cells (B). These two cell types were starved and then incubated with CXCL12 (50 ng/mL) alone or CXCL12 pre-incubated with JG6 (10, 50, or 100 μg/mL) for 15 min.
JG6 interacted with CXCL12
We next assessed how JG6 affects CXCL12/CXCR4 axis signaling. Previous studies in our laboratory have suggested that the structure of JG6 resembles the structure of GAGs and that GAGs appear to interact with CXCL12. We therefore utilized the SPR assay to identify the possible interaction between CXCL12 and JG6. JG6 was immobilized on NLC sensor chips. When CXCL12 was injected into the biosensor, a typical increase in SPR response corresponding to the association phase was observed in a dose-dependent manner (5–80 nmol/L). Upon displacement of the chemokine by the running buffer, a remarkable decrease in the SPR response signal emerged, consistent with the dissociation phase (Figure 4). The binding curve was fitted and demonstrated a multivalent binding mode with a clear association rate constant (kon) of 11.7 nmol/L and an equilibrium dissociation rate constant of 16.5 nmol/L. These data supported the findings above that indicate JG6 has a high affinity for CXCL12.

JG6 binds to CXCL12. (A) Binding response curve. (B) Equilibrium dissociation curve. Surface plasmon resonance (SPR) analysis of the interaction between JG6 and CXCL12. CXCL12 was injected at a concentration of 80, 40, 20, 10, 5 nmol/L (from top to bottom). The equilibrium dissociation constant was calculated by BIAcore software 3.1.
The automated docking simulation exemplified by JG6-CXCL12 interaction was performed to understand the interaction mode between them. The crystal structure of human CXCL12 was utilized as the target structure, whereas the disaccharide of JG6 was used as a docking probe. We found that JG6 docked well to CXCL12 with a low docking energy at -13.08 kcal/mol. JG6 appeared to interact with CXCL12 via multiple polar interactions, including 6 ionic bonds and 7 hydrogen bonds (Figure 5).

The interaction mode of JG6 and CXCL12 modeled by molecular docking. (A) The structure of JG6 in disaccharide form. (B) The interaction mode of JG6 and CXCL12. CXCL12 is shown in the white cartoon with JG6 shown as green sticks. The key residues from CXCL12 that interact with JG6 are also shown as sticks. Hydrogen bonds are indicated by red dashed lines, and ion bonds are indicated by yellow dashed lines.
These data, taken together, suggested that JG6 impaired the CXCL12/CXCR4 axis likely via a direct interaction with the chemokine CXCL12, which resulted in impaired intracellular signal transduction downstream of CXCL12 stimulation.
Discussion
The CXCL12/CXCR4 axis plays a critical role in cancer progression, including cell proliferation, survival, angiogenesis and particularly chemotactic metastasis. As a result, the CXCL12/CXCR4 axis has become a widely studied anti-cancer drug target. In this study, we have shown that JG6, a novel marine-derived oligosaccharide, interferes with the CXCL12/CXCR4 signaling axis and its associated biological functions in cancer cells. JG6 not only significantly reversed CXCL12-induced proliferation of cancer cells but also dramatically diminished CXCL12-mediated cell migration in vitro.
CXCL12 binds to GAGs, such as heparin sulfate (HS). CXCL12 binds to the S-domains of HS with high affinity predominantly through 2-O- and N-sulfate groups16. HS chains are linear repeating polymers composed of β-D-glucuronate and α-D-N-acetylglucosamine17 with a relatively uniform pattern of alternating sulfated and unmodified regions along the HS chain18. Previous studies of HS structure have identified a CXCL12-binding domain8,19. Moreover, a series of sulfated GAG mimetics targeting CXCL12 exhibited anti-cancer activity11. Based on the structural similarity between JG6 and GAGs, we hypothesized that JG6 was likely to bind to CXCL12. Using the SPR assay, we showed that JG6, like HS, directly binds to CXCL12 in a multivalent binding mode, which was consistent with the molecular docking model of the interaction. The calculated energy showed that JG6 can effectively bind to CXCL12 with a relatively low energy (-13.28 kcal/mol). Together, these results suggest that JG6 may directly interact with CXCL12, thereby blocking the CXCL12/CXCR4 axis and the subsequent biological functions such as tumor growth and migration.
In summary, our study shows that the structural features of JG6 enable it to interact with CXCL12 and provides an alternative for targeting the CXCL12/CXCR4 axis. The impact of JG6 on the CXCL12/CXCR4 axis may account for the anticancer activity of JG6.
Author contribution
Wei-wei WEN, Yi CHEN, and Jian DING designed the study; Wei-wei WEN performed experiments, analyzed the results and wrote the manuscript; Yi CHEN, Jian DING, and Mei-yu GENG revised the manuscript; Xian-ling XIN synthesized compounds; and Shao XIE was involved in part of the study.
References
Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev 2010; 29: 709–22.
Balkwill F . Cancer and the chemokine network. Nat Rev Cancer 2004; 4: 540–50.
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008; 13: 206–20.
Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A 2010; 107: 20009–14.
Liekens S, Schols D, Hatse S . CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr Pharm Des 2010; 16: 3903–20.
Patrussi L, Baldari CT . The CXCL12/CXCR4 axis as a therapeutic target in cancer and HIV-1 infection. Curr Med Chem 2011; 18: 497–512.
Brule S, Friand V, Sutton A, Baleux F, Gattegno L, Charnaux N . Glycosaminoglycans and syndecan-4 are involved in SDF-1/CXCL12-mediated invasion of human epitheloid carcinoma HeLa cells. Biochim Biophys Acta 2009; 1790: 1643–50.
Laguri C, Arenzana-Seisdedos F, Lortat-Jacob H . Relationships between glycosaminoglycan and receptor binding sites in chemokines-the CXCL12 example. Carbohydr Res 2008; 343: 2018–23.
Lortat-Jacob H . The molecular basis and functional implications of chemokine interactions with heparan sulphate. Curr Opin Struct Biol 2009; 19: 543–8.
Sutton A, Friand V, Brule-Donneger S, Chaigneau T, Ziol M, Sainte-Catherine O, et al. Stromal cell-derived factor-1/chemokine (C-X-C motif) ligand 12 stimulates human hepatoma cell growth, migration, and invasion. Mol Cancer Res 2007; 5: 21–33.
Friand V, Haddad O, Papy-Garcia D, Hlawaty H, Vassy R, Hamma-Kourbali Y, et al. Glycosaminoglycan mimetics inhibit SDF-1/CXCL12-mediated migration and invasion of human hepatoma cells. Glycobiology 2009; 19: 1511–24.
Zhang J, Chen Y, Xin XL, Li QN, Li M, Lin LP, et al. Oligomannurarate sulfate blocks tumor growth by inhibiting NF-kappaB activation. Acta Pharmacol Sin 2010; 31: 375–81.
Li QN, Liu HY, Xin XL, Pan QM, Wang L, Zhang J, et al. Marine-derived oligosaccharide sulfate (JG3) suppresses heparanase-driven cell adhesion events in heparanase over-expressing CHO-K1 cells. Acta Pharmacol Sin 2009; 30: 1033–8.
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004; 47: 1739–49.
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006; 49: 6177–96.
Sadir R, Imberty A, Baleux F, Lortat-Jacob H . Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J Biol Chem 2004; 279: 43854–60.
Salmivirta M, Lidholt K, Lindahl U . Heparan sulfate: a piece of information. FASEB J 1996; 10: 1270–9.
Murphy KJ, Merry CL, Lyon M, Thompson JE, Roberts IS, Gallagher JT . A new model for the domain structure of heparan sulfate based on the novel specificity of K5 lyase. J Biol Chem 2004; 279: 27239–45.
Rueda P, Balabanian K, Lagane B, Staropoli I, Chow K, Levoye A, et al. The CXCL12gamma chemokine displays unprecedented structural and functional properties that make it a paradigm of chemoattractant proteins. PLoS One 2008; 3: e2543.
Acknowledgements
The project was supported by the National Natural Science Foundation of China (No 81072666) and the National Basic Research Program Grant of China (No 2013CB932503).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wen, Ww., Xie, S., Xin, Xl. et al. Oligomannurarate sulfate inhibits CXCL12/SDF-1-mediated proliferation and invasion of human tumor cells in vitro. Acta Pharmacol Sin 34, 1554–1559 (2013). https://doi.org/10.1038/aps.2013.83
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.83
Keywords
This article is cited by
-
Angiosuppressive properties of marine-derived compounds—a mini review
Environmental Science and Pollution Research (2017)
-
CXCL12 expression promotes esophageal squamous cell carcinoma proliferation and worsens the prognosis
BMC Cancer (2016)
-
The B-RafV600E inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury
Cell Death & Disease (2014)
