
Abstract
Aim:
To establish a population pharmacokinetics (PPK) model for lamotrigine (LTG) in Chinese children with epilepsy in order to formulate an individualized dosage guideline.
Methods:
LTG steady-state plasma concentration data from therapeutic drug monitoring (TDM) were collected retrospectively from 284 patients, with a total of 404 plasma drug concentrations. LTG concentrations were determined using a HPLC method. The patients were divided into 2 groups: PPK model group (n=116) and PPK valid group (n=168). A PPK model of LTG was established with NONMEM based on the data from PPK model group according to a one-compartment model with first order absorption and elimination. To validate the basic and final model, the plasma drug concentrations of the patients in PPK model group and PPK valid group were predicted by the two models.
Results:
The final regression model for LTG was as follows: CL (L/h)=1.01*(TBW/27.87)0.635*e−0.753*VPA*e0.868*CBZ*e0.633*PB, Vd (L)= 16.7*(TBW/27.87). The final PPK model was demonstrated to be stable and effective in the prediction of serum LTG concentrations by an internal and external approach validation.
Conclusion:
A PPK model of LTG in Chinese children with epilepsy was successfully established with NONMEM. LTG concentrations can be predicted accurately by this model. The model may be very useful for establishing initial LTG dosage guidelines.
Similar content being viewed by others

Introduction
Lamotrigine (LTG) is a new antiepileptic drug and is chemically unrelated to any of the traditional drugs. Although it was initially used in association with other anticonvulsant drugs in the treatment of refractory epilepsy (EP), LTG monotherapy has recently been shown to be useful in the treatment of various types of epilepsy and epilepsy syndromes, both in children and adults1,2,3.
Although therapeutic drug monitoring (TDM) of LTG was not initially recommended, LTG use has increased due to high interindividual variability and the effects of certain antiepileptic drugs often associated with LTG, such as inhibitors (eg, valproic acid) or inducers (eg, carbamazepine, phenobarbital, phenytoin and primidone)4,5.
Pharmacokinetics (PK) is the science of determining the absorption, distribution, metabolism, and excretion of drugs in vivo. The traditional method for calculating individual PK parameters was to collect multiple (up to 7–10) blood samples from a single patient at different times after a single dose. This method was not always accepted by patients, especially children. Population pharmacokinetic analysis is an attractive approach because it uses fewer samples (as few as 1–2) from each patient. Only a few population PK analyses of LTG in children with epilepsy have been published6.
The objective of the current analysis is to establish a population pharmacokinetics (PPK) model for LTG in Chinese children with epilepsy to formulate an individualized dosage guideline.
Material and methods
Patients
The protocol for this study was approved by the Regional Ethics Committee, and consent was obtained from all of the children and their parents. The enrollment criteria were as follows: (1) the children were diagnosed as having epilepsy based on seizures and an electroencephalogram by a clinician; (2) the LTG therapy duration was over 1 month to ensure that the measured concentration was at a steady-state trough; and (3) patients had normal liver and kidney function. The exclusion criteria were as follows: (1) the patients did not have good compliance (good compliance defined as a patient who did not forget to take LTG or the co-medications even once for at least 1 month) and (2) having serious adverse drug reactions.
Sparse TDM data for LTG serum concentrations from 284 pediatric patients with EP at the Department of Pediatrics at Peking University First Hospital (Beijing, China) were collected. The patients were from all over the country and were taking LTG alone or concomitantly with other antiepileptic drugs (AEDs), such as carbamazepine, phenobarbital, valproic acid, oxcarbazepine, clonazepam, levetiracetam and topiramate.
The patients were divided into 2 groups: the PPK model group (n=116) and the PPK valid group (n=168). Table 1 summarizes the demographic and clinical characteristics of the patients included in the study.
Plasma sampling and serum concentration assay
Blood samples were drawn before breakfast and before taking drugs in the morning. A total of 404 plasma drug concentrations were collected approximately at the end of the dosing interval and in the steady-state situation (Css, min). One to ten plasma concentration time points were collected per patient.
The LTG serum concentrations were assayed using a high-performance liquid chromatographic technique with an ultraviolet detector (Chromsystems, Waters Company, Milford Massachusetts, USA). The mean±SD of the assay at the standard concentration points of 0.625, 1.25, 2.5, 5, 10, 20 and 40 μg/mL were 0.63±0.01, 1.22±0.02, 2.42±0.10, 4.82±0.10, 9.86±0.34, 20.08±0.45, and 41.92±1.68 μg/mL, respectively. Both intraday and interday variation coefficients were <6.5%.
Pharmacokinetic modeling
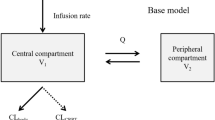
PPK analysis was performed with NONMEM-V (San Francisco, University of California, USA). A one-compartment, open kinetic model with first-order absorption and elimination (specified to NONMEM by the ADVAN2 and TRANS2 routines) was assumed according to the available bibliographic information7,8,9,10. Because the LTG steady-state trough plasma concentration data collected did not provide information about the extent and rate of absorption processes, the bioavailability (F) and absorption rate (Ka) were fixed at 1.0 and 1.0 h−1, respectively7. Thus, the PK parameters estimated were CL (the most important PK parameter in terms of optimizing drug dosage regimens) and Vd.
Exponential-error models were used to describe interindividual variability: CLj=CLpop*eηj, where CLj was values of CL for the jth patient; CLpop was the population mean CL; and ηj was the interindividual random variability with mean of zero and variances of ω2.
The intraindividual residual in concentration was modeled as: Cobsij=Cpredij*(1+ɛij), where Cobsij and Cpredij were the observed and predicted ith concentrations in the jth patient respectively, and ɛij was a random variable with a mean of zero and a variance of σ2.
The covariates of age, sex, total body weight (TBW) and concurrent therapy with VPA, OXC, CZP, LEV, TPM, and inducer antiepileptic drugs, such as CBZ and PB, were analyzed in the model. To elucidate the preliminary relationships between the CL obtained using a Bayesian maximum posteriori estimation (POSTHOC option in NONMEM) and covariates, a graphic approach to exploratory data analysis and the step-wise generalized additive model (GAM) implemented in Xpose were used11.
The inclusion of a fixed-effect parameter in the basic model quantifies the relationship between CL and a covariate and allows us to determine whether that covariate significantly improves the ability of the model to predict the observed concentration time profile. The criterion for estimation of statistical significance was a reduction in the minimum value of objective function (MOF) between these models. The objective function was defined as -2loglikelihood, and a minimal reduction of 3.84 (χ2=3.84 for P<0.05) for every extra parameter was necessary. Other diagnostic criteria for the retention of a covariate in the model were a reduction in unexplained interindividual variability for the associated PK parameter, an improvement in the graphic diagnostic model (evaluated by randomly distributed weighted residuals), a closer relationship between the predicted and observed concentrations, and the 95% confidence interval estimated using standard errors (SE) not including a zero value. In addition, the percentage estimation error of fixed and random parameters should not be higher than 25% and 50%, respectively12. Thus, the fully generated model was then subjected to backward elimination in which each model parameter was assigned a zero value using a more stringent criterion of statistical significance (χ2=7.88 for P<0.005).
Model validation
The PPK parameter values of LTG were calculated using NONMEM software with the data of 116 patients in the PPK model group. Subsequently, a basic and a final model were set up. The model validation included an internal validation (data of validation from the PPK model group) and an external validation (data of validation from the PPK valid group). To validate the basic and the final model, the drug concentrations from patients in the PPK model group and PPK valid group were predicted by the two models, respectively.
To validate the stability of the final model, the data from the PPK model group were divided into 10 groups randomly. Each group included 90% of the data from the PPK model group. The CLpop of each group was calculated by NONMEM software using the control file of the final model. Subsequently, each CLpop was fixed in the control file of the final model, and the ΔOFV (variety of objective function value between the valid group and the PPK model group) was calculated. The final model was stable if ΔOFV was <3.84 (χ2=3.84 for P<0.05).
Results
The initial model was as follows:

(θ1 was the population mean CL (L/h) estimated; θ6 was the population mean Vd (L) estimated; andηwas the interindividual random variability with mean of zero and variances of ω2).
A graphic exploratory analysis of the relationship between the individual Bayesian CL and the covariates analyzed by GAM revealed that age and TBW were clearly correlated with CL. Regarding the discrete variables analyzed and modeled as multiplicative factors, the effect of combined treatment with VPA elicited a decrease in CL, whereas inducers (CBZ, PB) were associated with an increase in this PK parameter. According to the graphic exploratory analysis, no other covariate was found to significantly improve the NONMEM fit. Thus, when sex and concomitant use of OXC, CZP, LEV, and TPM were included in the model, the magnitude of the estimates was negligible, and the percentage of associated SE was greater than 100%, indicating a lack of statistical and clinical significance of these covariates (Table 2). When a backward elimination was performed, only the effects of TBW and concomitant VPA, CBZ and PB on CL emerged as statistically significant covariates.
The final regressive model was as follows:

(θ1 was the population mean CL (L/h) estimated; θ2 was the fixed parameter relating to TBW; 27.87 (kg) was the average TBW; θ3 to θ5 were the fixed parameters relating to co-medication; θ6 was the population mean Vd (L) estimated; and η was the interindividual random variability with mean of zero and variances of ω2).
It was intended to be a fixed allometric covariate model such as Vd (L)=16.7*(WT/27.87), assuming 45 L is from an adult with 75 kg body weight; 16.7 was calculated by solving 45=θ6*(75/27.87).
In this model, the estimate of the CV for interindividual variability in CL was 25.8% (final model) versus 64.8% (basic model). In addition, the fixed-effect and random-effect parameters were estimated with an RSE of less than 25% and 50%, respectively (Table 3).
Figure 1 shows scatter plots of the measured LTG concentrations versus those predicted by the basic model (up) and the final model (down) (internal approach validation). These plots show the improvement in fit obtained with the latter model. Furthermore, the correlation coefficient of the linear regression between the observed versus predicted concentrations with the final model was 0.740, which is significantly better than 0.089 (basic model).

Observed versus predicted lamotrigine (LTG) plasma concentrations (population predicted concentration) according to basic (up) and final (down) population models (internal approach validation). Dotted line: regression lines.
Figure 2 shows scatter plots of the measured LTG concentrations versus those predicted by the basic model (up) and the final model (down) (external approach validation). These plots show the improvement in fit obtained with the latter model. Furthermore, the correlation coefficient of the linear regression between the observed versus predicted concentrations with the final model was 0.759, which is significantly better than 0.138 (basic model).

Observed versus predicted lamotrigine (LTG) plasma concentrations (population predicted concentration) according to basic (up) and final (down) population models (external approach validation). Dotted line: regression lines.
Table 4 shows that the final model was stable because the ΔOFV of ten valid groups were all under 3.84.
Discussion
The aim of this study was to quantitatively assess the effect of demographic and clinical variables on LTG PK in Chinese children with epilepsy. Despite LTG's wide clinical use, standard dosage regimens continue to be used even though they may often be suboptimal because of the high variability found in CL values in the target population7,10,13,14,15,16,17,18. This observation, together with previous studies that have established definable relationships between LTG blood concentrations and therapeutic and toxic effects19,20,21, justify TDM as an indicator of drug exposure for the individualization of LTG doses4,5. In this sense, the availability of a population kinetic model able to predict LTG concentrations for different situations is essential for the correct selection of the dosage schedules based on a Bayesian approach. However, only the initial dose may benefit from the population PK model because once the concentrations are measured, the future dose adjustment can be based on the observed concentration and the dose-proportional PK assumption.
Although several studies have addressed LTG PK5,7,8,9,10,13,14,15,16,22,23, only a few LTG studies using NONMEM have been conducted specifically in epileptic children. Consequently, the rationale for the design of the present study was to characterize LTG CL, the covariates affecting its variability, and unexplained residual and interindividual variabilities in Chinese epileptic children.
Our findings have suggested the following significant predictors for LTG clearance in the target population: total body weight, and co-medication with valproate and carbamazepine or phenobarbital.
Including the quantitative TBW covariate, LTG CL in the final model proposed was 0.039 L·h−1·kg−1, which is higher than the values of approximately 0.021 to 0.035 L·h−1·kg−1 that were reported in PK studies evaluating small numbers of adult patients and healthy adult volunteers using standard approaches (2-stage methods with a 1-compartment model)8,9,10,24. Our study has shown a nonlinear increase of the CL and Vd of LTG with an increase in total body weight. Similar results have been reported in pediatric and elderly populations of epileptic patients6,13,16.
The enzyme-inducing antiepileptic drugs associated with the treatment proved to have a significant effect on LTG CL. The induction effect of PB or CBZ was observed; thus, CL was increased by 1.9–2.4 fold compared to monotherapy. These results are similar to those from previous studies conducted in patients taking these drugs that also reported increased LTG CL with values ranging from 0.044 to 0.084 L·h−1·kg−17,25,26,27.
Co-medication with VPA has a significant reduction in CL by a factor of 0.47 compared with patients on LTG monotherapy. This observation is similar to the findings of previous studies, with values for this parameter ranging from 0.004 to 0.031 L·h−1·kg−1 and a mean of approximately of 0.015 L·h−1·kg−126,27,28,29,30,31.
When an inducing antiepileptic drug (PB or CBZ) and VPA are included in treatments with LTG, both effects, induction and inhibition, are balanced, and LTG CL shows little difference with respect to that observed in monotherapy.
The covariate analysis failed to detect a statistically significant effect of age on LTG CL, which is most likely due to the inclusion of TBW in the final model. In addition, TBW had a close correlation with age in children.
The covariates of sex and concomitant administration with other AEDs, including OXC, CZP, LEV, and TPM, also did not contribute to the variability in CL. Therefore, it is suggested that other AEDs did not influence this PK parameter.
The covariates included in the final model reduced the magnitude of the interindividual variability of LTG CL by 39% (25.8% vs 64.8%). This unexplained variability was of similar magnitude to those reported in earlier studies7,13,14,15,16,32,33,34.
Based on the final model proposed, Table 5 gives the predicted CL and the doses of LTG required to obtain a mean steady-state LTG concentration of 6 μg/mL for patients receiving LTG monotherapy or polytherapy. The magnitude of the interindividual and residual variabilities involves a broad range of expected LTG plasma concentrations for a fixed-dosage regimen, which could justify the TDM of LTG in epileptic patients.
The predictive performance of the model has been subjected to a preliminary evaluation in a total of 213 LTG concentrations from 168 epileptic children. These patients had demographics and clinical characteristics that were similar to those of the patients used to develop the model. Individual PK parameters were estimated using the population model, which was subsequently used to predict the LTG concentrations at the same times as those actually observed. These results show an acceptable accuracy.
Conclusions
Although these results are preliminary and not definite, the final PPK model was demonstrated to be stable and effective in the prediction of serum LTG concentrations by internal and external approach validation. Future use of this model will permit us to perform an external validation in a greater number of patients in order to prospectively evaluate its relevance in clinical practice.
References
Mullens EL . Clinical experience with lamotrigine monotherapy in adults with newly diagnosed epilepsy. Clin Drug Invest 1998; 16: 125–33.
Biton V, Sackellares JC, Vuong A, Hammer AE, Barrett PS, Messenheimer JA . Double-blind, placebo-controlled study of lamotrigine in primary generalized tonic-clonic seizures. Neurology 2005; 65: 1737–43.
Goa K, Ross SR, Chrisp P . Lamotrigine. A review of its pharmacological proper ties and clinical efficacy in epilepsy. Drugs 1993; 46: 152–76.
Chong E, Dupuis LL . Therapeutic drug monitoring of lamotrigine. Ann Pharmacother 2002; 36: 917–20.
Morris RG, Black AB, Harris AL, Batty AB, Sallustio BC . Lamotrigine and therapeutic drug monitoring: retrospective survey following the introduction of a routine service. Br J Clin Pharmacol 1998; 46: 547–51.
Chen C . Validation of a population pharmacokinetic model for adjunctive lamotrigine therapy in children. Br J Clin Pharmacol 2000; 50: 135–45.
Grasela TH, Fiedler-Kelly J, Cox E, Womble GP, Risner ME, Chen C . Population pharmacokinetics of lamotrigine adjunctive therapy in adults with epilepsy. J Clin Pharmacol 1999; 39: 373–84.
Cohen AF, Land GS, Breimer DD, Yuen WC, Winton C, Peck AW . Lamotrigine, a new anticonvulsant: pharmacokinetics in normal humans. Clin Pharmacol Ther 1987; 42: 535–41.
Rambeck B, Wolf P . Lamotrigine clinical pharmacokinetics. Clin Pharmacokinet 1993; 25: 433–43.
Perucca E . The clinical pharmacokinetics of the new antiepileptic drugs. Epilepsia 1999; 40 (Suppl 9): S7–S13.
Jonsson EN, Karlsson MO . Xpose — an S-PLUS based population pharmacokinetic/pharma codynamic model building aid for NONMEM. Comput Methods Programs Biomed 1999; 58: 51–64.
Vozeh S, Maitre PO, Stanski DR . Evaluation of population (NONMEM) pharmacokinetic parameter estimates. J Pharmacokinet Biopharm 1990; 18: 161–73.
Gidal BE, Anderson GD, Rutecki PR, Shaw R, Lanning A . Lack of an effect of valproate concentration on lamotrigine pharmacokinetics in developmentally disabled patients with epilepsy. Epilepsy Res 2000; 42: 23–31.
Hussein Z, Posner J . Population pharmacokinetics of lamotrigine monotherapy in patients with epilepsy: retrospective analysis of routine monitoring data. Br J Clin Pharmacol 1997; 43: 457–65.
Chan V, Morris RG, Ilett KF, Tett SE . Population pharmacokinetics of lamotrigine. Ther Drug Monit 2001; 23: 630–5.
Milovanovic JR, Jankovic SM . Population pharmacokinetics of lamotrigine in patients with epilepsy. Int J Clin Pharmacol Ther 2009; 47: 752–60.
Garnett WR . Lamotrigine pharmacokinetics. J Child Neurol 1997; 12: 10–5.
Armijo JA, Bravo J, Cuadrado A, Herranz JL . Lamotrigine serum concentration-to-dose ratio: influence of age and concomitant antiepileptic drug and dosage implications. Ther Drug Monit 1999; 21: 182–90.
Fröscher W, Keller F, Vogt H, Krämer G . Prospective study on concentration-efficacy and concentration-toxicity: correlations with lamotrigine serum levels. Epileptic Disord 2002; 4: 49–56.
Gidal BE, Welty TE . The concentration-effect relationship with lamotrigine (LTG). Epilepsia 1997; 38: 260.
Kilpatrick ES, Forrest G, Brodie MJ . Concentration-effect and concentration-toxicity relations with lamotrigine: a prospective study. Epilepsia 1996; 37: 534–8.
Biton V . Pharmacokinetics, toxicology and safety of lamotrigine in epilepsy. Expert Opin Drug Metab Toxicol 2006; 2: 1009–18.
Almeida AM, Falcão AC, Sales F, Baldeiras I, Rocha MJ, Caramona MM . Lamotrigine pharmacokinetic evaluation in epileptic patients submitted to VEEG monitoring. Eur J Clin Pharmacol 2006; 62: 737–42.
Posner J, Holdich T, Crome P . Comparison of lamotrigine pharmacokinetics in young and elderly healthy volunteers. J Pharm Med 1991; 1: 121–8.
Bialer M . The pharmacokinetics and interactions of new antiepileptic drugs: an overview. Ther Drug Monit 2005; 27: 722–6.
Weintraub D, Buchsbaum R, Resor SR Jr, Hirsch LJ . Effect of antiepileptic drug comedication on lamotrigine clearance. Arch Neurol 2005; 62: 1432–6.
Rambeck B, Specht U, Wolf P . Pharmacokinetic interactions of the new antiepileptic drugs. Clin Pharmacokinet 1996; 31: 309–24.
Bottiger Y, Svensson JO, Sthale L . Lamotrigine drug interactions in a TDM material. Ther Drug Monit 1999; 21: 171–4.
Kanner AM, Frey M . Adding valproate to lamotrigine: a study of their pharmacokinetic interaction. Neurology 2000; 55: 588–91.
Mataringa MI, May TW, Rambeck B . Does Lamotrigine influence valproate concentrations? Ther Drug Monit 2002; 24: 631–6.
Anderson GD, Yau MK, Gidal BE, Harris SJ, Levy RH, Lai AA, et al. Bidirectional interaction of valproate and lamotrigine in healthy subjects. Clin Pharmacol Ther 1996; 60: 145–56.
Sheiner LB, Beal SL . NONMEM Users Guide, Parts I–VI. San Francisco: NONMEM Project Group, University of California San Francisco, 1989.
Castel-Branco MM, Almeida AM, Falcão AC, Macedo TA, Caramona MM, Lopez FG . Lamotrigine analysis in biological fluids by high performance liquid chromatography (HPLC). J Chromatography B Biomed Sci Appl 2001; 755: 119–27.
Rivas N, Buelga DS, Elger CE, Santos-Borbujo J, Otero MJ, Domínguez-Gil A, et al. Population pharmacokinetics of lamotrigine with data from therapeutic drug monitoring in German and Spanish patients with epilepsy. Ther Drug Monit 2008; 30: 483–9.
Acknowledgements
The authors thank teacher Yun-xiu WANG, Dr De-chun JIANG, Li-ping ZHANG, Ying LIU, and Ying-hui WANG at Peking University First Hospital and teacher Hong-wen HU, Dr Xiu-yun YE, and Xu-lai SHI at Yuying Children′s Hospital Affiliated to Wenzhou Medical College, for their contributions to the study. This project was supported by the National Natural Science Foundation of China (No 81171630).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
He, Dk., Wang, L., Qin, J. et al. Population pharmacokinetics of lamotrigine in Chinese children with epilepsy. Acta Pharmacol Sin 33, 1417–1423 (2012). https://doi.org/10.1038/aps.2012.118
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.118
Keywords
This article is cited by
-
Effect of Age-Related Factors on the Pharmacokinetics of Lamotrigine and Potential Implications for Maintenance Dose Optimisation in Future Clinical Trials
Clinical Pharmacokinetics (2018)
-
Authors’ Reply to Standing et al.: “Effect of Age-Related Factors on the Pharmacokinetics of Lamotrigine and Potential Implications for Maintenance Dose Optimisation in Future Clinical Trials”
Clinical Pharmacokinetics (2018)
-
Population pharmacokinetics of lamotrigine co-administered with valproic acid in Chinese epileptic children using nonlinear mixed effects modeling
European Journal of Clinical Pharmacology (2018)
-
Population pharmacokinetic models of lamotrigine in different age groups of Chinese children with epilepsy
European Journal of Clinical Pharmacology (2017)
-
Clinical Pharmacokinetics of New-Generation Antiepileptic Drugs at the Extremes of Age: An Update
Clinical Pharmacokinetics (2013)
