
Abstract
Obstructive sleep apnea (OSA) is well known for its metabolic as well as neurobehavioral consequences. Chronic intermittent hypoxia (IH) is a major component of OSA. In recent years, substantial advances have been made in elucidating the cellular and molecular mechanisms underlying the effect of chronic IH on neurocognitive functions, many of which are based on studies in animal models. A number of hypotheses have been put forward to explain chronic IH-induced neurological dysfunctions. Among these, the roles of oxidative stress and apoptosis-related neural injury are widely accepted. Here, focusing on results derived from animal studies, we highlight a possible role of reduced expression of brain-derived neurotrophic factor (BDNF) in causing impairment in long-term synaptic plasticity and neurocognitive functions during chronic IH. The possible relationship between BDNF and previous findings on this subject will be elucidated.
Similar content being viewed by others

Introduction
Obstructive sleep apnea (OSA), a very common breathing and sleep disorder, is associated with intermittent hypoxia (IH) resulting from upper airway obstruction of structural or neural causes1, 2. The most distinct features of OSA are episodes of oxyhemoglobin desaturations, which are terminated by brief periods of microarousals that could lead to sleep deprivation, fragmentation and alteration in sleep pattern3. During OSA, arterial O2 saturation could drop to very low level (50%–60%) within every cycle. This problem is alarmingly common and likely to be over-looked by the general population. The prevalence in men and women have been estimated to be 24% and 9% respectively if we only assess the frequency of increased obstructive events during sleep4. A large variety of problems are associated with OSA, including cardiovascular morbidity, hypertension, obesity, dyslipidemia, insulin resistance, and neurocognitive malfunctions5, 6. Because of the prevalence of OSA, there is a substantial cost that the society has to pay for their treatment, and also the failure in their diagnosis. In addition, OSA incurs society costs in the form of reduced work efficiency, occupational and motor vehicle accidents, decreased quality of life and morbidity. Statistical analysis confirmed that treatment reduces the medical cost of OSA7, highlighting the significance and impact of improving available treatment strategies.
OSA and neurological functions
It is well known that OSA is not just a breathing disorder with metabolic consequences. The cyclic hypoxia and sleep fragmentation could lead to impaired brain functions which severely degrade performance in daily lives and work, and is one of the major causes of sleepiness and concentration-deficit related traffic accidents. It is well known that OSA results in cognitive deficits including decreased attention and vigilance, phonological problem, irritability, impairment in executive functions and long-term memory8, 9, 10, 11, 12, 13, 14, 15. However, very little is known about the detailed events happening in the central nervous system in OSA subjects, and the relative roles of sleep fragmentation and intermittent hypoxia. Furthermore, although surgery and continuous positive airway pressure (CPAP) are useful treatments for OSA, whether long-term changes happening in the brain could be reverted is not known. These are important questions to be addressed and the answers are just beginning to be unraveled.
Chronic IH-induced impairment in memory and neuroplasticity
There are a large number of studies in human OSA subjects to investigate the origin of the neurocognitive problems, many of which are based on brain imaging. Techniques such as structural magnetic resonance imaging and proton magnetic resonance spectroscopy revealed significant changes in various brain regions and metabolism in OSA patients15. It should be pointed out that, because of the simultaneous occurrence of intermittent hypoxemia and sleep fragmentation in OSA, dissecting the influences of these two factors on cognitive functions, and what aspects of cognition, are difficult in human subjects. Understandably, these techniques are limited in providing mechanistic explanation of the pathological events at the cellular level. On the other hand, based on animal models, attempts and significant advances have been made in the last decade in unveiling the relationship between OSA and cognitive dysfunction and the underlying mechanisms.
To assess the neurobehavioral effects of episodic hypoxia in the absence of sleep fragmentation, Gozal and colleagues in early years established an animal model to study the anatomical and behavioral correlates of chronic episodic hypoxia in the rat11, 13. They established that exposure to IH during sleep cycle of adult rats is associated with significant spatial learning deficits as well as increased neuronal loss within susceptible brain regions such as the hippocampus and cortex. Subsequent studies have confirmed that chronic IH treatment, as well as sleep fragmentation, as models of OSA, could impair spatial memory functions of rodents to different degrees, as measured by the conventional water maze tests16, 17, 18, 19, 20, 21, 22.
There are a number of factors and pathways that have been proposed to account for the effects of OSA-associated IH and sleep disturbance on neurocognitive functions. An early notion asserted that episodes of hypoxia could trigger apoptosis programs in neurons in areas including the hippocampus and cortex, and could lead to cytoarchitectural disorganization11, 16. In fact, apoptosis in the hippocampal CA1 region could be detected as early as 1–2 d in the IH-treated rats, preceding the appearance of memory deficits11. Consistent with this idea, a significant number of hippocampal slices obtained from the hypoxic animals failed to exhibit tetanus-induced potentiation of populations spikes, measured at 15 min post-stimulation23. The effects on the conventional early phase (ie up to 1 h) and late-phase (longer than 3 h) long-term potentiation (LTP) were however not addressed in this study.
The relatively mild degree of apoptosis detected in the brains of the IH animals11 raises the question of whether apoptosis could explain the neurocognitive malfunctions of the animals. In fact, it is possible that chronic IH can cause a general compromise of oxidative phosphorylation and consequently poor maintenance of ion gradients of neurons. In other words, the physiological function of the neurons may be compromised before explicit apoptosis. There were few attempts to examine the direct effects of chronic IH on the excitability of hippocampal neurons, and their synaptic transmission, in the animal model of OSA. Nevertheless, it has been shown that in the developing nervous system, IH will affect neuronal excitability and its maturation by altering the expression of Na channels and ion transporters24, 25. In the adult mice, we also found that chronic IH decreases membrane input resistance and excitability of hippocampal CA1 neurons26.
The reasons for the compromised neuronal function or apoptosis in the hippocampus or other brain areas are not entirely known. However, it is highly probable that oxidative stress plays a significant role (reviewed by WANG et al27). Thus, it has been well established that there are increased expressions of oxidative stress markers found in the brains of rodents subjected to IH treatment28, 29, 30. Administration of anti-oxidants28 or over-expressing superoxide dismutase30 attenuated reactive oxygen species (ROS) production and apoptosis in chronic IH-treated animals. More recent evidence supports a specific role of NADPH oxidase in IH-induced oxidative stress31, 32. However, up to now, the source and mechanism of ROS generation and its impact on neurocognitive deficits in IH are not entirely clear.
There exist other possible mechanisms by which IH could affect the hippocampus and therefore learning and memory behaviours. For example, FUNG et al33 suggested that intermittent hypoxia produces abnormally high level of glutamate and causes excitotoxicity in hippocampal neurons. LI et al34 concluded from their study that intermittent hypoxia in the rat is associated with an increased expression of iNOS which may play a critical role in IH-mediated neurobehavioural deficits. Furthermore, inflammation, which has been shown to play important roles in mediating the peripheral effects of IH35, may contribute to neural injury in the brain36.
It is well known that the hippocampal circuit is critical for the formation of spatial memory. However, a causal relationship between chronic IH and hippocampal synaptic plasticity has not been established until very recently. In a recent study based on a mouse model of OSA, we showed for the first time that there was a significant decrease in early phase long-term potentiation (E-LTP) in the hippocampi of both 7-d and 14-d IH-treated mice, while there was no apparent effect on the 3-d IH group26. This result provides an explanation for the well-documented memory deficits associated with OSA and its models. Of importance, it was demonstrated that not only the conventional E-LTP was impaired by IH, but the late-phase LTP (L-LTP), which better correlates with the formation of long-term memory, was also impaired and to a more significant extent. However, whether those factors that have been proposed to contribute to cognitive dysfunction, namely oxidative stress, apoptosis, decreased neuronal excitability, excitotoxicity, inflammatory response etc, is a cause of impaired LTP has not been demonstrated.
Critical role of decreased BDNF expression in chronic intermittent hypoxia
Brain-derived neurotrophic factor (BDNF), as a member of the neurotrophin family, plays key roles in neuronal survival and differentiation during development37, 38, 39. BDNF is also known to be expressed and released in an activity-dependent manner in the central nervous system and can acutely modulate synaptic transmission and plasticity40, 41, 42, 43. In a previous study, we and co-workers reported that BDNF is critical in the expression of L-LTP in the hippocampus suggesting that it is a key protein in long-term memory formation44. Consistent with this idea, a single amino acid polymorphism in the BDNF gene has been shown to affect the cortical morphology and memory in human45, 46. In addition, the conversion of proBDNF to mature form of BDNF is tightly regulated by central tissue plasminogen activator (tPA) which catalyses the conversion of plasminogen to plasmin. Plasmin then cleaves proBDNF to mature BDNF.
In our study on the chronic IH mouse model26, we found that the expression of BDNF was reduced significantly after chronic IH treatment. Compelling evidence for critical role of BDNF was provided by showing that exogenous application or surgical replenishment of BDNF by intraventricular injection could rescue and prevent, respectively, IH-induced LTP deficits. Thus, BDNF could be a crucial factor contributing to the absence of normal hippocampal plasticity and therefore memory function in the IH model.
At present, the exact reason causing BDNF decrease in chronic IH is unknown. Being a neurotrophic factor, the level of BDNF has been shown to be increased under some pathological conditions of the brain47, 48 and spinal cord49. This is usually regarded as a compensatory mechanism by the nervous system to help boost the survival of neurons. However, prolonged insult such as chronic IH may compromise the ability of neurons, and probably astrocytes as well, to express BDNF. The time-dependent decrease in BDNF level we found in our chronic IH model is in line with this notion. Interestingly, the level of another neurotrophic factor, NT4/5, was not decreased (unpublished data) indicating that the effect of IH on BDNF is specific, and also argues against the possibility that the decrease in BDNF level is simply due to neuronal loss. It is known that chronic IH affects gene transcription, including those driven by CREB16. While the total CREB production remains unchanged, the phosphorylated form of CREB was reduced, maximally at 3 d, after hypoxic treatment. Since BDNF is a CREB-dependent gene product, the impact of chronic IH at the gene level could provide an explanation of the observed decrease in BDNF expression. On the other hand, we found that the expression of plasmin, the extracellular enzyme that helps to cleave pro-BDNF to mature forms of BDNF is reduced in the chronic IH model (unpublished data). This result is consistent with our observation that the pro-BDNF level is not significantly affected by chronic IH treatment, and points to a role of proteolytic cleavage of proBDNF to mature BDNF rather than transcription of BDNF gene.
It should be pointed out that, in our study, although we did not specifically induce sleep fragmentation or deprivation on the subjects, the loss or interference in sleep could contribute to a certain extent the observed changes in BDNF as the sleep architecture may be affected50, 51. Furthermore, a recent study employing a chronic IH paradigm with a much longer cycle length resulted in an enhancement of BDNF expression in the hippocampus52, and in another study, a less severe paradigm of daily intermittent hypoxia also augments BDNF expression in the spinal cord53. These results suggest that the number and frequency of hypoxia/re-oxygenation cycles could be a major factor in determining the effect on BDNF expression.
Relationship between BDNF, oxidative stress, and apoptosis
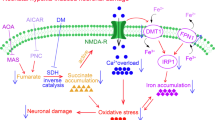
As described above, oxidative stress induced by repeated hypoxia/re-oxygenation challenge and ROS-induced apoptosis are two closely-related factors that are widely accepted to contribute to neuronal damage under chronic IH condition27. Given our discovery of the importance of BDNF in restoring hippocampal functions in IH animals, how can we reconcile our findings with these hypotheses? There is growing evidence that neurotrophic factors such as BDNF can significantly prevent neuronal damage caused by oxidative stress, as in neurodegenerative diseases (reviewed by NUMAKAWA et al54), or as shown in in vitro cultures against ROS generation and action directly55. Thus, it is possible that lack of BDNF in chronic IH not only contributes to impaired long-term synaptic plasticity but also fails to prevent neuronal injury, including apoptosis, induced by ROS. In other words, lack of BDNF is a key factor in the cascades of events leading to neurocognitive deficits in OSA, as depicted in Figure 1. In this model, chronic IH could lead directly to decrease in neuronal excitability, BDNF level and the generation of ROS. These factors act together in a synergistic manner to increase apoptosis and also impairment in long-term synaptic plasticity underlying memory function. Obviously, further experiments are needed to scrutinize this hypothesis.

Proposed interactions between BDNF reduction and other pathological processes that lead to neuronal injury and decreased neuroplasticity in OSA. Chronic intermittent hypoxia and/or sleep fragmentation leads directly to decrease in neuronal excitability, decrease in BDNF expression and generation of ROS. These factors act together in a synergistic manner to increase apoptosis and also impairment in long-term synaptic plasticity underlying memory function. In this model, decreased expression of BDNF plays a pivotal role in ROS generation, apoptosis as well as impairment in synaptic plasticity.
Future directions
Here we would like to highlight a few issues that interest us and at the same time that we feel are important. First, the establishment of the chronic IH model in mimicking the hypoxia/re-oxygenation cycles in OSA has advanced our understanding of the pathophysiology of OSA. However, the differential effects of IH on short-term working memory and long-term memory are far from clear. In fact, it has been suggested that sleep fragmentation has a selective effect on working memory function56. Thus, the impact of chronic IH vs sleep disturbance on different types of memory, as well as the involvement of BDNF level in these processes, are key issues that need to be addressed.
IH definitely affects neuronal functions, but despite the obvious importance, the question of exactly what happens to neuronal activities in different brain regions during IH has never been addressed. Previous attempts only relied on in vitro brain slice preparations and recordings could be made only after the animals had been sacrificed26, 57. Long-term recording of the firing properties of neurons in vivo during or after the IH will give a direct answer to this question and will provide big insight into the cause of cognitive dysfunctions in OSA.
One of the main thrusts in investigating the mechanisms of synaptic plasticity impairment and neurocognitive deficits in OSA models is to provide a scientific basis for potential pharmacological treatment. We have shown that multiple intraventricular injections of BDNF is beneficial to IH-induced LTP impairment in the mice model26. Therefore, the level of BDNF may be a novel therapeutic target to improve OSA-associated neurocognitive impairments. There were reports indicating that endogenous BDNF level could be elevated by short-term administration of specific compounds, for example, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid AMPA receptor modulator ampakine58. Ampakines are a group of small molecules that can delay deactivation and reduce desensitization of AMPA type glutamate receptors and thereby increase the size and duration of ligand-gated current flow, enhancing glutamate transmission59, 60. In addition, previous studies showed that regular ampakine administration would increase the expression of BDNF. Considering that ampakines can also cross the blood-brain barrier, are bioactive orally and improve cognitive function without obvious side effects, they are of great potential and in fact have been partially proved to be candidate for a range of neurological disability and disturbances, including Alzheimer's disease and Huntington's disease58, 61, 62, 63. We hypothesize that ampakines are beneficial to the neurocognitive problems found in OSA by its action in elevating endogenous BDNF level in the brain. If proven, adjunct pharmacological treatment for cognitive problems in OSA would become feasible.
References
Caples SM, Gami AS, Somers VK . Obstructive sleep apnea. Ann Intern med 2005; 142: 187–97.
Mathieu A, Mazza S, De'cary A, Massicotte-Marquez J, Petit D, Gosselin N, et al. Effects of obstructive sleep apnea on cognitive function: A comparison between younger and older OSAS patient. Sleep Med 2007; 9: 112–20.
Deegan PC, McNicholas WT . Pathophysiology of obstructive sleep apnea. Eur Respir J 1995; 8: 1161–78.
Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S . The occurrence of sleep-disordered breathing among middle-aged adults. N Eng J Med 1993; 328: 1230–5.
Lavie L . Oxidative stress — a unifying paradigm in obstructive sleep apnea and comorbidities. Prog Cardiovas Dis 2009; 4: 303–12.
Levy P, Bonsignore MR, Eckel J . Sleep, sleep-disordered breathing and metabolic consequences. Eur Respir J 2009; 34: 243–60.
Hoffman B, Wingenbach D, Kagey AN, Schaneman JL, Kasper D . The long-term health plan and disability cost benefit of obstructive sleep apnea treatment in a commercial motor vehicle driver population. J Occup Environ Med 2010; 52: 473–7.
Ali NJ, Pitson D, Stradling JR . Sleep disordered breathing: effects of adenotonsillectomy on behaviour and psychological functioning. Eur J Pediatr 1996; 155: 56–62.
Chervin RD, Dillon JE, Bassetti C, Ganoczy DA, Pituch KJ . Symptoms of sleep disorders, inattention, and hyperactivity in children. Sleep 1997; 20: 1185–92.
Gozal D . Sleep-disordered breathing and school performance in children. Pediatrics 1998; 102: 616–20.
Gozal D, Daniel JM, Dohanich GP . Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci 2001; 21: 2442–50.
Gozal D . Effects of intermittent hypoxia on neurological function. In Brain Hypoxia and Ischemia 2009; (Eds Haddad GG, Yu SP). Humana Press.
Row BW, Kheirandish L, Neville JJ, Gozal D . Impaired spatial learning and hyperactivity in developing rats exposed to intermittent hypoxia. Pediatr Res 2002; 52: 449–53.
Tsai JC . Neurological and neurobehavioral sequelae of obstructive sleep apnea. NeuroRehabilitation 2010; 26: 85–94.
Jackson ML, Howard ME, Barnes M . Cognition and daytime functioning in sleep-related breathing disorders. Prog Brain Res 2011; 190: 53–68.
Goldbart A, Row BW, Kheirandish L, Schurr A, Gozal E, Guo SZ, et al. Intermittent hypoxic exposure during light phase induces changes in cAMP response element binding protein activity in the rat CA1 hippocampal region: Water maze performance correlates. Neuroscience 2003; 122: 583–90.
Goldbart A, Cheng ZJ, Brittia KR, Gozal D . Intermittent hypoxia induces time-dependent changes in the protein kinase B signaling pathway in the hippocampal CA1 region of the rat. Neurobiol Dis 2003; 14: 440–6.
Gozal D, Row BW, Gozal E, Kheirandish K, Neville JJ, Brittian KR, et al. Temporal aspects of spatial task performance during intermittent hypoxia in the rat: evidence for neurogenesis. Eur J Neurosci 2003; 18: 2335–42.
Kheirandish K, Gozal D, Pequignot JM, Pequignot J, Row BW . Intermittent hypoxia during development induces long-term alteration in spatial working memory, monoamines, and dendritic branching in rat frontal cortex. Pediatr Res 2005; 58: 594–9.
Kheirandish K, Row BW, Li RC, Brittian KR, Gozal D . Apolipoprotein E-deficient mice exhibit increased vulnerability to intermittent hypoxia-induced spatial learning deficits. Sleep 2005; 28: 1412–7.
Tartar JL, Ward CP, McKenna JT, Thakkar M, Arrigoni E, McCarley RW, et al. Hippocampal synaptic plasticity and spatial learning are impaired in a rat model of sleep fragmentation. Eur J Neurosci 2006; 23: 2739–48.
Ward CP, McCoy JG, McKenna JT, Connolly NP, McCarley RW, Strecker RE . Spatial learning and memory deficits following exposure to 24 h of sleep fragmentation or intermittent hypoxia in a rat model of obstructive sleep apnea. Brain Res 2009; 1294: 128–37.
Payne RS, Goldbart A, Gozal D, Schurr A . Effect of intermittent hypoxia on long-term potentiation in rat hippocampal slices. Brain Res 2004; 1029: 195–9.
Gu XQ, Haddad GG . Maturation of neuronal excitability in hippocampal neurons of mice chronically exposed to cyclic hypoxia. Am J Physiol Cell Physiol 2003; 284: C1156–63.
Zhao P, Xue J, Gu XQ, Haddad GG, Xia Y . Intermittent hypoxia modulates Na+ Channel expression in developing mouse brain. Int J Dev Neurosci 2005; 23: 327–33.
Xie H, Leung KL, Chen L, Chan YS, Cheung PK, Fok TF, et al. Brain-derived neurotrophic factor rescues and prevents chronic intermittent hypoxia-induced impairment of hippocampal long-term synaptic plasticity. Neurobiol Dis 2010; 40: 155–62.
Wang Y, Zhang SXL, Gozal D . Reactive oxygen species and the brain in sleep apnea. Respir Physio Neurobiol 2010; 174: 307–16.
Row BW, Liu R, Xu W, Kheirandish L, Gozal D . Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am J Respir Crit Care Med 2003; 167: 1548–53.
Zhan G, Fenik P, Pratico D, Veasey SC . Inducible nitric oxide synthase in long-term intermittent hypoxia: hypersomnolence and bran injury. Am J Respir Crit Care Med 2005; 171: 1414–20.
Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, et al. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience 2004; 126: 313–23.
Hui-guo L, Kui L, Yan-ning Z, Yong-jian X . Apocynin attenuate spatial learning eficits and oxidative responses to intermittent hypoxia. Sleep Med 2010; 11: 205–12.
Nair D, Dayyat EA, Zhang SX, Wang Y, Gozal D . Intermittent hypoxia-induced cognitive deficits are mediated by NADPH oxidase activity in a murine model of sleep apnea. PLoS One 2011; 6: e19847.
Fung SL, Xi MC, Zhang JH, Sampogna S, Yamuy J, Morales FR, et al. Apnea promotes glutamate-induced excitotoxicity in hippocampal neurons. Brain Res 2007; 1179: 42–50.
Li RC, Row BW, Kheirandish L, Brittian KR, Gozal E, Guo SZ, et al. Nitric oxide synthase and intermittent hypoxia-induced spatial learning deficits in the rat. Neurobiol Dis 2004; 17: 44–53.
Ryan S, McNicholas WT, Taylor C . A critical role for p38 map kinase in NF-kB signaling during intermittent hypoxia/reoxygenation. Biochem Biophys Res Com 2007; 355: 728–33.
Li RC, Row BW, Gozal E, Kheirandish L, Fan Q, Brittian KR, et al. Cyclooxygenase 2 and intermittent hypoxia-induced spatial deficits in the rat. Am J Respir Crit Care Med 2003; 15: 469–75.
Thoenen H . The changing scene of neurotrophic factors. Trends Neurosci 1991; 14: 165–70.
Lewin GR, Barde YA . Physiology of the neurotrophins. Ann Rev Neurosci 1996; 19: 289–317.
Bibel M, Barde YA . Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 2000; 14: 2919–37.
Poo MM . Neurotrophin as synaptic modulators Nat. Rev Neurosci 2001; 2: 24–32.
Lu B . Acute and long-term synaptic modulation by neurotrophins. Prog Brain Res 2004; 146: 137–50.
Rose CR, Blum R, Kafitz KW, Kovalchuk Y, Konnerth A . From modulator to mediator: rapid effects of BDNF on ion channels. Bioessays 2004; 26: 1185–94.
Lu Y, Christian K, Lu B . BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem 2008; 89: 312–23.
Pang P, Teng H, Zaitsev E, Woo NT, Sakata K, Zhen S, et al. Proteolytic conversion from pro- to mature BDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 2004; 305: 487–91.
Egan M, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003; 112: 257–69.
Pezawas L, Verchinski BA, Mattay VS, Callicott JH, Kolachana BS, Straub RE, et al. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci 2004; 24: 10099–102.
Chiaretti A, Pastra M, Polidori G, Di Rocco C, Caresta E, Antonell A, et al. Correlation between neurotrophic factor expression and outcome of children with severe traumatic brain injury. Intensive Care Med 2003; 29: 1329–38.
Chiaretti A, Antonelli A, Piastra M, Genovese O, Polidori G, Aloe L . Expression of neurotrophic factors in cerebrospinal fluid and plasma of children with viral and bacterial meningoencephalitis. Acta Paediatr 2004; 93: 1178–84.
Dale-Nagle EA, Hoffman MS, MacFarlane PM, Satriotomo I, Lovett-Barr MR, Vinit S, et al. Spinal plasticity following intermittent hypoxia: implications for spinal injury. Ann N Y Acad Sci 2010; 1198: 252–9.
Veasey SC, Davis CW, Fenik P, Zhan G, Hsu YJ, Pratico D, et al. Long-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain region. Sleep 2004; 27: 194–201.
Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, et al. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med 2006; 7: 7–16.
Zhu XH, Yan HC, Zhang J, Qu HD, Qiu XS, Chen L, et al. Intermittent hypoxia promotes hippocampal neurogenesis and produces antidepressant-like effects in adult rats. J Neurosci 2010; 30: 12653–63.
Wilkerson JE, Mitchell GS . Daily intermittent hypoxia augments spinal BDNF levels, ERK phosphorylation and respiratory long-term facilitation. Exp Neurol 2009; 217: 116–23.
Numakawa T, Matsumoto T, Numakawa Y, Ricahrds M, Yamawaki S, Kunugi H . Protective action of neurotrophic factors and estrogen against oxidative stress-mediated neurodegeneration. J Toxicol 2011; 2011: 405194.
Boutahar N, Reynaud E, Lassabliere F, Borg J . Brain-derived neurotrophic factor inhibits cell cycle reentry but not endoplasmic reticulum stress in cultured neurons following oxidative or excitotoxic stress. J Neurosci Res 2010; 8: 2263–71.
Thomas RJ, Rosen BR, Stern CE, Weiss W, Wong KK . Functional imaging of working memory in obstructive sleep-disordered nreathing. J Appl Physiol 2005; 98: 2226–34.
Tartar JL, McKenna JT, Ward CP, McCarley RW, Strecker RE, Brown RE . Sleep fragmentation reduces hippocampal CA1 pyramidal cell excitability and response to adenopsine. Neurosci Lett 2010; 469: 1–5.
Simmons DA, Rex CS, Palmer L, Pandyaranjan V, Fedulov V, Gall CM, et al. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington's disease knockin mice. Proc Natl Acad Sci U S A 2009; 106: 4906–11.
Arai AC, Kessler M . Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr Drug Targets 2007; 8: 583–602.
Nagarajan N, Quast C, Boxall AR, Shahid M, Rosenmund C . Mechanism and impact of allosteric AMPA receptor modulation by the ampakine CX546. Neuropharmacology 2001; 41: 650–63.
Kramar EA, Chen LY, Lauterborn JC, Simmons DA, Gall CM, Lynch G . BDNF upregulation rescues synaptic plasticity in middle-aged ovariectomized rats. Neurobiol Aging 2010. doi:10.1016/j.neurobiolaging.2010.06.008
Lynch G . Memory enhancement: the search for mechanism-based drugs. Nat Neurosci 2002; 5: 1035–8.
Wezenberg E, Verkes RJ, Ruigt GS, Hulstjin W, Sabbe BG . Acute effects of ampakine farampator on memory and information processing in healthy elderly volunteers. Neuropsychopharmacol 2007; 32: 1272–83.
Acknowledgements
The authors' work described in this article was supported by the Research Grants Council of Hong Kong, China (Project No 478308).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xie, H., Yung, Wh. Chronic intermittent hypoxia-induced deficits in synaptic plasticity and neurocognitive functions: a role for brain-derived neurotrophic factor. Acta Pharmacol Sin 33, 5–10 (2012). https://doi.org/10.1038/aps.2011.184
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.184
Keywords
This article is cited by
-
Association between brain-derived neurotrophic factor levels and obstructive sleep apnea: a systematic review and meta-analysis
Sleep and Breathing (2023)
-
Correlation between cognitive impairment and serum markers in patients with obstructive sleep apnea–hypopnea syndrome
Sleep and Breathing (2023)
-
Role of Oxidative Stress in the Occurrence and Development of Cognitive Dysfunction in Patients with Obstructive Sleep Apnea Syndrome
Molecular Neurobiology (2023)
-
7,8-Dihydroxyflavone protects retinal ganglion cells against chronic intermittent hypoxia-induced oxidative stress damage via activation of the BDNF/TrkB signaling pathway
Sleep and Breathing (2022)
-
Impact of histaminergic H3 receptor antagonist on hypoglossal nucleus in chronic intermittent hypoxia conditions
Psychopharmacology (2021)
