
Abstract
Amyloid β (Aβ) peptides have long been viewed as a potential target for Alzheimer's disease (AD). Aggregation of Aβ peptides in the brain tissue is believed to be an exclusively pathological process. Therefore, blocking the initial stages of Aβ peptide aggregation with small molecules could hold considerable promise as the starting point for the development of new therapies for AD. Recent rapid progresses in our understanding of toxic amyloid assembly provide a fresh impetus for this interesting approach. Here, we discuss the problems, challenges and new concepts in targeting Aβ peptides.
Similar content being viewed by others

Introduction
More than 100 years have passed since Alzheimer's disease (AD) was first characterized. However, due to the lack of effective treatment, AD remains pandemic in the 21st century, imposing enormous social, and economic burdens on patients and their families1. Modern demographic trends compound the problem; our aging global population has led to a steep increase in the number of individuals with AD. In the United States alone, more than 13 million individuals are predicted to be afflicted with AD by the year 20502, leading to an overburden of scarce healthcare resources. Some studies estimate that the present availability of a treatment that can delay disease onset by 6.7 years would decrease the prevalence of AD 38% by 20503. Such disease-modifying treatments would lower the annual cost of individual patient care by up to $24 000, thereby reducing the national cost of AD by trillions of dollars through the year 20504, 5.
Unfortunately, currently available treatments, eg, Aricept and Memantine, usually provide at best only temporary and incomplete symptomatic relief. The marginal benefits provided by current therapies emphasize the urgent need to develop effective disease-modifying AD treatments.
Amyloid β–targeting strategies
Disease-modifying strategies currently being pursued for AD mainly focus on two AD-related proteins, amyloid β (Aβ) and Tau. Of these, Aβ has attracted the most attention by far.
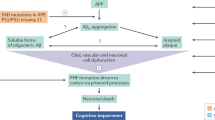
Substantial data derived from genetics, animal modeling, and biochemical studies support the idea that Aβ, the major component of senile plaques, plays a central role in AD pathophysiology6, 7. Paradoxically, Aβ peptides — the seeds of AD — are present at birth, and these neurotoxic peptides are continuously produced throughout life. Thus, the most direct target in anti-Aβ therapy is reduction of Aβ production, which has led naturally to a focus on β- and γ-secretase inhibitors8, 9, 10, 11, 12, 13. However, despite considerable effort on this front, few novel chemical compounds based on this strategy have reached clinic trials. Safety issues are the overriding reason; because γ-secretase has many physiologically essential substrates, undesirable side effects are inevitable. One such substrate is the Notch signaling protein, which plays an intermediate and essential role in the differentiation and proliferation of many cell types. The consequences of γ-secretase inhibition include impaired lymphocyte differentiation and altered intestinal goblet cell structure14, 15, 16. Thus, although the use of small-molecule γ-secretase modulators is receiving increasing attention as a promising therapeutic approach17, many companies have abandoned γ-secretase as a potential target.
In theory, inhibiting β-secretase should not carry the same risk of toxicity as γ-secretase inhibition. However, designing β-secretase inhibitors has been challenging. The β-secretase protein contains a large catalytic pocket; thus, the β-secretase inhibitors that have been developed to date are too large to penetrate the blood-brain barrier18, 19, 20. There may also be conceptual drawbacks to targeting β-secretase. The development of Aβ secretase inhibitors is based on the hypothesis that Aβ deposition in the brain is due to overproduction or poor clearance of Aβ. However, this is likely only true for cases of familial AD caused by genetic mutation. Non-genetic AD cases, which represent the majority of AD patients, do not carry mutations and do not necessarily have overproduction of amyloid precursor protein (APP)21. More importantly, a deeper understanding of Aβ has revealed that Aβ isoforms also serve as endogenous positive regulators of release probability at hippocampal synapses, with some studies suggesting that monomeric Aβ is beneficial for neurons22, 23. Thus, because Aβ production may be important for physiological health, inhibiting Aβ generation may not necessarily be a sound strategy. An alternative tactic is to focus on Aβ clearance. One approach is to enhance the peripheral Aβ “sink action” by sequestering plasma Aβ, for example using Aβ immunotherapy. The AN1792 Aβ vaccine was designed for this purpose; however, it failed in Phase II clinical trials due to the development of meningoencephalitis24. Nevertheless, it might be possible to circumvent such complications by targeting the proteolytic machinery in the brain. However, from a therapeutic point of view, it remains to be seen whether any of the relevant proteases will serve as a viable AD drug target25.
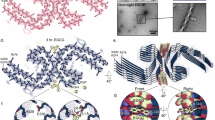
Aβ can aggregate into fibrils and oligomers (Figure 1). It was originally believed that only the large fibrils constituting mature neuritic amyloid plaques are toxic. However, in recent years research focus has shifted towards Aβ oligomers. Currently, Aβ peptide aggregation into toxic, prefibrillar oligomers is considered the key pathogenic event in the onset of AD26, 27. Consistent with this view, Aβ oligomers can: i) directly induce synaptic dysfunction and neuronal death, both which are responsible for AD initiation and progression28, 29, 30; and ii) trigger events such as oxidative damage and inflammation, which contribute to the progression of AD31, 32, 33. Although generation of Aβ monomers is, in and of itself, a physiologically relevant event, their aggregation into oligomers and fibrils is pathogenic. Thus, blocking aggregation (while sparing Aβ generation) should not lead to mechanism-based toxicity. Hence, the strategy of inhibiting aggregation of Aβ, more specifically the Aβ42 isoform, has emerged as a valid disease-modifying therapy for AD34.

The plaque formation.
Problems associated with inhibiting Aβ aggregation
Tremendous effort has been expended in recent years on developing small molecules that are capable of inhibiting Aβ aggregation. However, to date, no compounds have entered into clinical use. This is because the inhibition of Aβ aggregation requires blocking interactions between Aβ monomers, and historically, protein–protein interaction interfaces are particularly difficult drug targets. Thus, inhibiting Aβ aggregation has been deemed intractable35, 36. The challenges associated with targeting Aβ aggregation are substantial, with the most obvious obstacle being the sheer size and geometry of the protein interaction surface. First, the regions of protein–protein interactions are approximately 1500–3000 Å237, 38, 39, 40, whereas protein–small molecule interaction regions are only about 300–1000 Å2 41, 42. Thus, small molecules are often unable to produce sufficient steric hindrance to inhibit Aβ aggregation43. Second, the regions of protein–protein interactions are often relatively featureless, devoid of grooves or pockets into which a small molecule could dock in an energetically favorable manner43, 44, 45, 46, 47. The binding energy that drives protein–protein contacts is typically distributed over a large area that lacks a defined “hotspot” for pharmacological intervention48. Third, the highly plastic nature of protein surfaces can accommodate small molecules, thereby thwarting inhibition48. Together, these problems place severe restrictions on the development of Aβ aggregation inhibitors.
Designing solutions to these problems
To tackle these challenges, researchers have envisioned a Trojan horse strategy in which a small bifunctional molecule gains access to the relevant biological compartment, binds tightly to a chaperone, and thereby provides the necessary steric bulk to disrupt protein–protein interactions48. A second approach is to directly target the important chaperones, such as metals, in the brain. A growing body of evidence suggests that the presence of excess zinc, iron, copper and/or aluminum within senile plaques exacerbates Aβ-mediated oxidative damage and acts as a catalyst for Aβ aggregation in AD49. Thus, chelation therapies aimed at disrupting aberrant interactions between Aβ and metals have attracted considerable attention; one such example is PBT2, which is currently in Phase IIa clinical trials50. Unfortunately, to date, no new compounds employing this strategy have entered into clinical use.
The discovery that Aβ fibril formation is controlled by specific amino acids within the Aβ peptide itself has given renewed life to the idea that small molecules might occlude Aβ aggregation. Studies have demonstrated that various Aβ peptide regions contribute differently to Aβ aggregation and have identified important interactions among specific peptide regions that control this process51. For instance, the N-terminus52, 53, hydrophobic core54, 55, hinge or turn regions51, 56, 57 and C-terminus58, 59 of Aβ41-42 are all crucial for the peptide's ability to aggregate and promote neurotoxicity.
The importance of the His13–Lys16 (HHQK) region in oligomerization, fibril propagation and neurotoxicity is well known60, 61, 62. This four-residue region is also an essential component of the heparin-binding site for glycosaminoglycans (GAGs), which assist the HHQK motif in effecting a change in the Aβ secondary structure from a soluble, unordered α-helix to a stable β-sheet–rich conformation. These β-sheet–rich structures, in turn, have an affinity for associating with other monomers to form oligomers, protofibrils and fibrils that aggregate into plaques60. The β-hairpin structure serves to nucleate the folding of Aβ monomers, which is the rate-limiting step in fibril formation56, 57, 63. The formation of hinge or turn regions brings together two hydrophobic segments in space and produces the β-strand structure. In this region, Lys28 forms a salt bridge with Asp23/Glu22, which further stabilizes the structure63, 64, 65. Electrostatic and hydrophobic interactions between these non-native side chains bring the peptides together, allowing native backbone−backbone interactions to initiate the assembly of β-sheets, albeit in non-native β-sheet registries64. The hydrophobic segments are the main core of Aβ aggregates, and hydrophobic interactions are an important driving force in Aβ aggregate formation66, 67. Based on these observations, a β-sheet breaker was first investigated as a potential inhibitor of Aβ aggregation68. A substantial body of evidence suggests that the C-terminus of Aβ42 plays a key role in controlling oligomerization; indeed, several studies of prefibrillar Aβ have suggested that the C-terminus of Aβ42 is more rigid than the C-terminus of the more abundant but less toxic Aβ4069, 70, 71. This increased rigidity has been attributed to interactions involving C-terminal residues Ile41 and Ala42, which stabilize a putative turn conformation71. Although these subregions contribute differently to structure formation, each is important for Aβ aggregation.
By targeting these subregions for binding, a small molecule will be more likely to directly occlude Aβ aggregation. In fact, most inhibitors that have entered clinical trials target these specific subregions within the Aβ sequence (Figure 2). Tramiprosate (3-aminopropanesulfonic acid), a mimic of GAGs, targets the HHQK subregion at the N-terminus. Although the structural simplicity of tramiprosate makes it highly specific to Aβ, it ultimately failed in the late stages of a Phase III clinical trial72. Despite this setback, the data obtained provided evidence that inhibitors targeting specific Aβ subregions represent a viable approach for AD treatment73. The agents targeting C-terminus feature one scaffold, the cyclohexanehexol stereoisomers. Among these, scyllo-inositol, which interferes with Aβ fibril assembly by competing with endogenous phosphatidylinositol for binding to Aβ41-4259, has shown promise in current Phase II trials. The hydrophobic central region is yet another promising target for Aβ fibrillation inhibitors, which include RS-040674.

The subregion-targets in Aβ.
The quest for small molecules that target Aβ
As the strategy of inhibiting Aβ aggregation has increasingly gained acceptance, greater numbers of inhibitors have been developed and the structure-activity relationships of potent inhibitors have been systematically explored75. These studies revealed that typical Aβ aggregation inhibitors such as Congo red (CR), chrysamine G (CG) and curcumin share a similar chemical scaffold. These molecules contain two aromatic groups or inositol groups (with a suitable substituted group) separated by a backbone of the appropriate length75. We speculate that the two terminal groups interact with Aβ protein residues to provide the binding affinity, whereas the linker facilitates binding of inhibitors to specific subregions. Determining which subregion the inhibitors target will be key to future research efforts. A molecule that targets a specific subregion should satisfy the following design criteria: (1) it should contain terminal groups suitable for interacting with residues in, or adjacent to, the subregion; and (2) the linker should be of an appropriate length to join the two terminal groups, span the subregion, and provide sufficient steric hindrance. Molecules designed according to these specifications would therefore be capable of inhibiting Aβ aggregation by binding to a specific subregion.
The handful of inhibitors that have been developed to date do not possess this chemical scaffold. Scyllo-inositol contains only one terminal group and can therefore target only a very small region. Fortunately, scyllo-inositol targets the C-terminus, which is important for Aβ aggregation. However, the presence of a single terminal group restricts the area of the targeted subregion to the C-terminus, and the lack of a linker prevents scyllo-inositol from modulating other targeted subregions. Additionally, a single terminal group results in a weaker binding avidity. In the case of tramiprosate, its simple structure provides specificity for Aβ, yet, for unknown reasons it failed in Phase III clinical trials72. Thus, although these atypical molecules possess inhibitory activity, they are severely limited by their weak binding affinity and small occupied region.
To our knowledge, most inhibitors target only one subregion. In fact, if inhibitors could target multiple specific subregions in concert, they might act synergistically to effectively inhibit Aβ aggregation. Thus, in our view, the ideal Aβ-targeting inhibitor should be of appropriate length and suitable flexibility, and should contain multiple groups that interact with Aβ residues. These multiple groups would both convey tighter binding avidity and permit targeting of multiple subregions. In addition, having sufficient length and flexibility would enable the inhibitor to capture the entire Aβ peptide and accommodate conformational changes in the target. Unfortunately, our current lack of the Aβ peptide's detailed molecular structure precludes designing such inhibitors. The successful creation of these new inhibitors will require gaining a better understanding of the modules necessary for activity as well as the key elements in the amyloid surface required for aggregation and toxicity. Alternatively, it might also be possible to employ a combination of multiple molecules.
Future perspectives
Compounds such as peptide-based inhibitors, antibodies and small molecules that target specific Aβ subregions represent the first generation of amyloid-based therapeutics with the potential to demonstrate disease-modifying activity. Although the results of ongoing clinical trials are inconclusive, these compounds hold the promise of a new day in the development of disease-modifying therapies for AD; some Aβ aggregation inhibitors are listed in Table 1. As we gain additional insights into amyloid biology and AD itself, this will likely guide the development of the next generation of inhibitors.
References
Wimo A, Winblad B, Jonsson L . The worldwide societal costs of dementia: Estimates for 2009. Alzheimers Dement 2010; 6: 98–103.
Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA . Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003; 60: 1119–22.
Sloane PD, Zimmerman S, Suchindran C, Reed P, Wang L, Boustani M, et al. The public health impact of Alzheimer's disease, 2000–2050: potential implication of treatment advances. Annu Rev Public Health 2002; 23: 213–31.
Zhu CW, Torgan R, Scarmeas N, Albert M, Brandt J, Blacker D, et al. Home health and informal care utilization and costs over time in Alzheimer's disease. Home Health Care Serv Q 2008; 27: 1–20.
Leon J, Cheng CK, Neumann PJ . Alzheimer's disease care: costs and potential savings. Health Aff (Millwood) 1998; 17: 206–16.
Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G . Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci U S A 1991; 88: 7552–6.
Blanquet V, Goldgaber D, Turleau C, Creau-Goldberg N, Delabar J, Sinet PM, et al. The beta amyloid protein (AD-AP) cDNA hybridizes in normal and Alzheimer individuals near the interface of 21q21 and q22.1. Ann Genet 1987; 30: 68–9.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999; 286: 735–41.
Vassar R, Citron M . Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron 2000; 27: 419–22.
Gao Y, Pimplikar SW . The gamma-secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc Natl Acad Sci U S A 2001; 98: 14979–84.
Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y . Alzheimer's beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci U S A 2001; 98: 13554–9.
Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, et al. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 2001; 4: 231–2.
Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 2003; 9: 3–4.
van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005; 435: 959–63.
Tarassishin L, Yin YI, Bassit B, Li YM . Processing of Notch and amyloid precursor protein by gamma-secretase is spatially distinct. Proc Natl Acad Sci U S A 2004; 101: 17050–5.
Leissring MA, Murphy MP, Mead TR, Akbari Y, Sugarman MC, Jannatipour M, et al. A physiologic signaling role for the gamma-secretase-derived intracellular fragment of APP. Proc Natl Acad Sci U S A 2002; 99: 4697–702.
Tomita T . At the frontline of Alzheimer's disease treatment: gamma-secretase inhibitor/modulator mechanism. Naunyn Schmiedebergs Arch Pharmacol 2008; 377: 295–300.
Citron M . Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci 2004; 5: 677–85.
Opar A . Mixed results for disease-modification strategies for Alzheimer's disease. Nat Rev Drug Discov 2008; 7: 717–8.
Citron M . Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov 2010; 9: 387–98.
Nishitomi K, Sakaguchi G, Horikoshi Y, Gray AJ, Maeda M, Hirata-Fukae C, et al. BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice. J Neurochem 2006; 99: 1555–63.
Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, et al. Beta-amyloid monomers are neuroprotective. J Neurosci 2009; 29: 10582–7.
Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I . Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 2009; 12: 1567–76.
Lemere CA, Masliah E . Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol 2010; 6: 108–19.
De Strooper B . Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev 2010; 90: 465–94.
Resende R, Ferreiro E, Pereira C, Resende de Oliveira C . Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 2008; 155: 725–37.
Walsh DM, Selkoe DJ . Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 2004; 44: 181–93.
Reddy PH, Beal MF . Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med 2008; 14: 45–53.
Hu NW, Smith IM, Walsh DM, Rowan MJ . Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain 2008; 131: 2414–24.
Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R . Synaptic memory mechanisms: Alzheimer's disease amyloid beta-peptide-induced dysfunction. Biochem Soc Trans 2007; 35: 1219–23.
Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T . Inflammation in Alzheimer's disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 2009; 87: 181–94.
Ruan L, Kang Z, Pei G, Le Y . Amyloid deposition and inflammation in APPswe/PS1dE9 mouse model of Alzheimer's disease. Curr Alzheimer Res 2009; 6: 531–40.
Ali-Khan Z, Sipe JD, Du T, Riml H . Echinococcus multilocularis: relationship between persistent inflammation, serum amyloid A protein response and amyloidosis in four mouse strains. Exp Parasitol 1988; 67: 334–45.
Estrada LD, Soto C . Disrupting beta-amyloid aggregation for Alzheimer disease treatment. Curr Top Med Chem 2007; 7: 115–26.
Whitty A, Kumaravel G . Between a rock and a hard place? Nat Chem Biol 2006; 2: 112–8.
Hajduk PJ, Burns DJ . Integration of NMR and high-throughput screening. Comb Chem High Throughput Screen 2002; 5: 613–21.
Lo Conte L, Chothia C, Janin J . The atomic structure of protein-protein recognition sites. J Mol Biol 1999; 285: 2177–98.
Jones S, Thornton JM . Principles of protein-protein interactions. Proc Natl Acad Sci U S A 1996; 93: 13–20.
Teichmann SA . Principles of protein-protein interactions. Bioinformatics 2002; 18: S249.
Keskin O, Gursoy A, Ma B, Nussinov R . Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem Rev 2008; 108: 1225–44.
Smith RD, Hu L, Falkner JA, Benson ML, Nerothin JP, Carlson HA . Exploring protein-ligand recognition with Binding MOAD. J Mol Graph Model 2006; 24: 414–25.
Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol 2007; 25: 71–5.
Wells JA, McClendon CL . Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007; 450: 1001–9.
Arkin MR, Wells JA . Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov 2004; 3: 301–17.
Plewczynski D, Rychlewski L . Meta-basic estimates the size of druggable human genome. J Mol Model 2009; 15: 695–9.
Hambly K, Danzer J, Muskal S, Debe DA . Interrogating the druggable genome with structural informatics. Mol Divers 2006; 10: 273–81.
Hopkins AL, Groom CR . The druggable genome. Nat Rev Drug Discov 2002; 1: 727–30.
Gestwicki JE, Crabtree GR, Graef IA . Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science 2004; 306: 865–9.
Dong J, Canfield JM, Mehta AK, Shokes JE, Tian B, Childers WS, et al. Engineering metal ion coordination to regulate amyloid fibril assembly and toxicity. Proc Natl Acad Sci U S A 2007; 104: 13313–8.
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, et al. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008; 59: 43–55.
Maji SK, Ogorzalek Loo RR, Inayathullah M, Spring SM, Vollers SS, Condron MM, et al. Amino acid position-specific contributions to amyloid beta-protein oligomerization. J Biol Chem 2009; 284: 23580–91.
Gardberg AS, Dice LT, Ou S, Rich RL, Helmbrecht E, Ko J, et al. Molecular basis for passive immunotherapy of Alzheimer's disease. Proc Natl Acad Sci U S A 2007; 104: 15659–64.
McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, et al. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4-10 and inhibit cytotoxicity and fibrillogenesis. Nat Med 2002; 8: 1263–9.
Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH . Amyloid fibrils of the HET-s (218–289) prion form a beta solenoid with a triangular hydrophobic core. Science 2008; 319: 1523–6.
Permanne B, Adessi C, Saborio GP, Fraga S, Frossard MJ, Van Dorpe J, et al. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer's disease by treatment with a beta-sheet breaker peptide. FASEB J 2002; 16: 860–2.
Hoyer W, Gronwall C, Jonsson A, Stahl S, Hard T . Stabilization of a beta-hairpin in monomeric Alzheimer's amyloid-beta peptide inhibits amyloid formation. Proc Natl Acad Sci U S A 2008; 105: 5099–104.
Fawzi NL, Phillips AH, Ruscio JZ, Doucleff M, Wemmer DE, Head-Gordon T . Structure and dynamics of the Abeta (21–30) peptide from the interplay of NMR experiments and molecular simulations. J Am Chem Soc 2008; 130: 6145–58.
Fradinger EA, Monien BH, Urbanc B, Lomakin A, Tan M, Li H, et al. C-terminal peptides coassemble into Abeta42 oligomers and protect neurons against Abeta42-induced neurotoxicity. Proc Natl Acad Sci U S A 2008; 105: 14175–80.
McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MH, Phinney AL, et al. Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat Med 2006; 12: 801–8.
Bartolini M, Bertucci C, Bolognesi ML, Cavalli A, Melchiorre C, Andrisano V . Insight into the kinetic of amyloid beta (1–42) peptide self-aggregation: elucidation of inhibitors' mechanism of action. Chembiochem 2007; 8: 2152–61.
Williamson MP, Suzuki Y, Bourne NT, Asakura T . Binding of amyloid beta-peptide to ganglioside micelles is dependent on histidine-13. Biochem J 2006; 397: 483–90.
Watson DJ, Lander AD, Selkoe DJ . Heparin-binding properties of the amyloidogenic peptides Abeta and amylin. Dependence on aggregation state and inhibition by Congo red. J Biol Chem 1997; 272: 31617–24.
Melquiond A, Dong X, Mousseau N, Derreumaux P . Role of the region 23–28 in Abeta fibril formation: insights from simulations of the monomers and dimers of Alzheimer's peptides Abeta40 and Abeta42. Curr Alzheimer Res 2008; 5: 244–50.
Buchete NV, Hummer G . Structure and dynamics of parallel beta-sheets, hydrophobic core, and loops in Alzheimer's A beta fibrils. Biophys J 2007; 92: 3032–9.
Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC . Abeta40-Lactam (D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry 2005; 44: 6003–14.
Lu X, Wintrode PL, Surewicz WK . Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Natl Acad Sci U S A 2007; 104: 1510–5.
Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y . Evidence of fibril-like beta-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's beta-amyloid. Nat Struct Mol Biol 2007; 14: 1157–64.
Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B . Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat Med 1998; 4: 822–6.
Murakami K, Irie K, Ohigashi H, Hara H, Nagao M, Shimizu T, et al. Formation and stabilization model of the 42-mer Abeta radical: implications for the long-lasting oxidative stress in Alzheimer's disease. J Am Chem Soc 2005; 127: 15168–74.
Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB . On the nucleation of amyloid beta-protein monomer folding. Protein Sci 2005; 14: 1581–96.
Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, et al. In silico study of amyloid beta-protein folding and oligomerization. Proc Natl Acad Sci U S A 2004; 101: 17345–50.
Rauk A . Why is the amyloid beta peptide of Alzheimer's disease neurotoxic? Dalton Trans 2008; 10: 1273–82.
Blazer LL, Neubig RR . Small molecule protein-protein interaction inhibitors as CNS therapeutic agents: current progress and future hurdles. Neuropsychopharmacology 2009; 34: 126–41.
Nakagami Y, Nishimura S, Murasugi T, Kaneko I, Meguro M, Marumoto S, et al. A novel beta-sheet breaker, RS-0406, reverses amyloid beta-induced cytotoxicity and impairment of long-term potentiation in vitro. Br J Pharmacol 2002; 137: 676–82.
Reinke AA, Gestwicki JE . Structure-activity relationships of amyloid beta-aggregation inhibitors based on curcumin: influence of linker length and flexibility. Chem Biol Drug Des 2007; 70: 206–15.
Acknowledgements
This work was funded by grants from the National Natural Science Foundation of China (No 30472029) and the National Natural Science Foundation of China for Distinguished Young Scholars (No 30725046).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nie, Q., Du, Xg. & Geng, My. Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer's disease. Acta Pharmacol Sin 32, 545–551 (2011). https://doi.org/10.1038/aps.2011.14
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.14
Keywords
This article is cited by
-
Disaggregation of amyloid-beta fibrils via natural metabolites using long timescale replica exchange molecular dynamics simulation studies
Journal of Molecular Modeling (2024)
-
Human cystatin C induces the disaggregation process of selected amyloid beta peptides: a structural and kinetic view
Scientific Reports (2023)
-
Identification of potential inhibitors against Alzheimer-related proteins in Cordyceps militaris ethanol extract: experimental evidence and computational analyses
3 Biotech (2023)
-
Protein clearance strategies for disease intervention
Journal of Neural Transmission (2022)
-
Free radical biology in neurological manifestations: mechanisms to therapeutics interventions
Environmental Science and Pollution Research (2022)
