
Abstract
Aim:
To investigate whether down-regulation of peroxiredoxin 1 (Prx1) and/or peroxiredoxin 5 (Prx5) sensitizes human esophageal cancer cells to ionizing radiation (IR).
Methods:
Human esophageal carcinoma cell lines Eca-109 and TE-1 were used. Prx mRNA expression profiles in Eca-109 and TE-1 cells were determined using RT-PCR. Two highly expressed isoforms of Prxs, Prx1 and Prx5, were silenced by RNA interference (RNAi). Following IR, intracellular reactive oxygen species (ROS) and apoptosis were measured using flow cytometry, the activities of catalase, superoxide dismutase and glutathione peroxidase were measured, and the radiosensitizing effect of RNAi was observed. Tumor xenograft model was also used to examine the radiosensitizing effect of RNAi in vivo.
Results:
Down-regulation of Prx1 and/or Prx5 by RNAi does not alter the activities of catalase, superoxide dismutase and glutathione peroxidase, but made human tumor cells more sensitive to IR-induced apoptosis both in vitro and in vivo. When the two isoforms were decreased simultaneously, intracellular ROS and apoptosis significantly increased after IR.
Conclusion:
Silencing Prx1 and/or Prx5 by RNAi sensitizes human Eca-109 and TE-1 cells to IR, and the intracellular ROS accumulation may contribute to the radiosensitizing effect of the RNAi.
Similar content being viewed by others

Introduction
Over the past three decades, esophageal squamous cell carcinoma remains the dominant histological type of esophageal cancer in Europe and Asia. Despite technological advances in cancer therapies, the overall 5-year survival rate of this malignancy has remained at about 10% since 1980s1. Approximately 50%–70% of all cancer patients received radiotherapy during their treatment. However, except for those with very early-stage disease, radiation has had little impact on long-term survival2. Epidemiological data strongly suggest that the integration of radiation therapy into multimodal management approaches improves the outcome of therapy for esophageal cancer, but clinical radiation resistance continues to be a major problem3.
We hypothesize that this resistance is due, in part, to altered levels of antioxidant enzymes. It is well known that the therapeutic effect of ionizing radiation (IR) is damage of DNA and proteins caused by radiation and IR-induced intracellular reactive oxygen species (ROS). Among the antitumor effects of IR, ROS are crucial for cell survival4, 5. The enhanced constitutive oxidative stress renders tumor cells highly dependent on endogenous antioxidants, such as catalase (CAT), glutathione peroxidase (GPx), Cu/Zn-superoxide dismutase (SOD), Mn-SOD, and peroxiredoxins (Prxs). These endogenous antioxidants protect tumor cells from continuous intracellular ROS injury.
Among intracellular antioxidant defense enzymes, Prxs have been recently characterized as a group of thiol-containing proteins with efficient antioxidant capacity, and have been proved to play a critical role in peroxide detoxification6, 7. Altogether six distinct Prx isoforms have been identified in mammalian tissues, and elevated expression of different Prx isoforms has been documented in numerous malignancies8, 9, 10, 11. Furthermore, silencing of Prx expression is currently used to enhance the radiotherapy effect in the cells of breast cancer, lung carcinoma, intestinal cancer and colon cancer12.
A comparative proteomic analysis showed that the expression of Prx1, increased in human esophageal squamous cell carcinoma tissues as compared with adjacent normal tissues9. However, the Prx expression profile is not clear, as well as the relationship between Prx expression and human esophageal squamous cell radiosensitivity.
Therefore, we conducted the present study to get a clear picture of Prx expression. Meanwhile, we explored the influence of Prxs on the radiosensitivity of two human esophageal cancer cell lines, namely, Eca-109 and TE-1.
Materials and methods
Cell culture
Human esophageal carcinoma cell lines Eca-109 and TE-1 were obtained from Cell Bank, Chinese Academy of Sciences (Shanghai, China). The cell lines were cultivated at 37 °C in complete Dulbecco's modified Eagle's medium (DMEM, Invitrogen, USA) supplemented with 10% fetal calf serum (FCS, Gibco, USA), and 100 units/mL penicillin G, 100 mg/mL streptomycin sulfate, in a humidified atmosphere consisting of 95% air and 5% CO2.
Animals
Male BALB/c nude mice (4-6 weeks old) were used. All mice were handled according to the Guidelines for the Care and Use of Laboratory Animals. This study was approved by the Institutional Animal Care and Use Committee of Xi'an Jiaotong University, Xi'an, China.
Irradiation
For the investigation of radiosensitivity, IR was performed at a fixed dose rate of 0.262 Gy/min and a focus-surface distance of 100 cm from a linear accelerator (ELEKTA, Britain) located in the First Affiliated Hospital, School of Medicine, Xi'an Jiaotong University, Xi'an, China.
Profiling of Prx mRNA expression
Total RNA was isolated from cultured Eca-109 and TE-1 cells using RNAfast200 Kit (Fastagen Biotech, Shanghai, China), and first strand cDNA was synthesized from the total RNA (2 μg) using random hexamer primers and RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, USA); both kits were used according to the manufacturer's instructions. Quantitative real-time PCR was performed using SYBR Premix Ex Taq™ (Takara, Japan). Amplification and detection were performed using the ABI PRISM 7300 Sequence Detection System starting with 2 μL of cDNA. Profiling of Prx mRNA expression was detected by real-time PCR. The primer pairs (listed in Table 1) were synthesized by Sangon Biotech Co, Ltd (Shanghai, China). Real-time PCR was performed following the method taken by Thomas et al13. Beta-actin was used as an internal control.
Prx1 small interfering RNA (siRNA) transfection
RNA interference (RNAi) of Prx1 was performed using siRNA duplexes chemically synthesized by GenePharma Co, Ltd (Shanghai, China). The sense strand nucleotide sequence for Prx1 siRNA was 5′-CUGGAAACCUGGCAGUGAUTT-3′, and the anti-sense sequence was 5′-AUCACUGCCAGGUUUCCAGTT-3′. The negative control siRNA (siNeg) sense sequence was 5′-UUCUCCGAACGUGUCACGUTT-3′, while the antisense sequence was 5′-ACGUGACACGUUCGGAGAATT-3′. The siRNA duplexes were introduced into the cells using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's guidelines, and Prx1 knock-down was evaluated by Western blotting with antibodies to Prx1.
Prx5 small hairpin RNA (shRNA) transfection and isolation of clones stably expressing Prx5 shRNA
Prx5 specific shRNA (PGPU6/Neo-Prx5) and control shRNA vector (PGPU6/Neo-shNC) were purchased from Genepharma (Shanghai, China). The targeted sequence of PGPU6/Neo-Prx5 was GGAATCGACG-TCTCAAGAGGT, and the targeted sequence of the negative control PGPU6/Neo-shNC was GTTCTCCGAACGTGTCACGT. Eca-109 and TE-1 cells grown on 24-well plates were transfected either with PGPU6/Neo-Prx5 or negative control plasmid. Transfection was performed using Lipofectamine 2000. The cells were incubated at 37 °C in 5% CO2 overnight, and then DMEM plus 10% FCS was added. After another 12-h incubation the cells were re-plated at 1/10−1/40 dilution onto the 6-well plates. Selection with G418 (500 μg/mL) started on the next day and the process lasted for 2 weeks. Eca-109 and TE-1 clones with stably decreased expression of Prx5 as well as the control clones were obtained for further study. Prx5 knock-down was evaluated by Western blotting with antibodies to Prx5.
Western blot analysis
The protein expression of Prxs in Eca-109 and TE-1 cells were examined by Western blotting 72 h post transfection. The cells were washed twice in phosphate buffered saline (PBS), lysed in ice-cold radioimmune precipitation (RIPA) buffer, and then centrifuged for 10 min at 4 °C. Supernatant was collected and protein concentrations were determined and adjusted to 2 mg/mL using the Bio-Rad kit (Bio-Rad Laboratories, Hercules, CA, USA). Cell lysates were mixed with 3×Laemmli buffer and heated for 5 min at 95 °C. They were then resolved by SDS-PAGE (8% or 10% polyacrylamide gels), and transferred to polyvinylidene difluoride membranes (Immobilon™; Millipore Corp, Bedford, MA, USA) by electroblotting. The membranes were blotted with 10% nonfat milk, washed in Tris-buffered saline (TBS) Tween and incubated with primary rabbit polyclonal antibodies overnight at 4 °C. Washed with TBS Tween again, they were incubated with secondary antibody solution (horseradish peroxidase conjugated IgG) for 60 min at room temperature. The membranes were washed again, and then fluorescence detection was conducted using the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The following primary antibodies were used at dilutions: anti-Prx1, 1:1000; anti-Prx2, 1:1000; anti-Prx5, 1:1000; anti-β-actin, 1:800; secondary antibodies were used at dilutions of 1:5000. Protein loading equivalence was assessed by the expression of β-actin.
Measurement of enzymatic activities of CAT, GPx, Cu/Zn-SOD and Mn-SOD after RNAi
Eca-109 and TE-1 cells, according to the way they were treated, were divided respectively into 7 groups as follows: Blank group; Prx1 siNeg group, cells transfected with negative control siRNA; Prx1 siRNA group, cells transfected with Prx1 siRNA; Prx5 shNeg group, cells stably expressing a non-targeted control shRNA; Prx5 shRNA group, cells stably expressing Prx5 shRNA; Combined negative group (Prx5 shNeg and Prx1 siNeg), cells stably expressing a non-targeted control shRNA and transfected with Prx1 siNeg; Combined group (Prx5 shRNA and Prx1 siRNA), cells stably expressing Prx5 shRNA and transfected with Prx1 siRNA. With Eca-109 and TE-1 cells, we got altogether 14 cell groups. After transfection, cells were grown on T-25 flasks for 48 h at 37 °C. Before IR, cell lysates were centrifuged at 12 000×g for 10 min at 4 °C and the supernatant was collected for measuring the activities of CAT, GPx, Cu/Zn-SOD and Mn-SOD, respectively by a Catalase Analysis Kit, a Cellular Glutathione Peroxidase Assay Kit, and Cu/Zn-SOD and Mn-SOD Assay Kit with WST-1 (Beytime Institute of Biotechnology, China). The measurements were performed according to the manufacturer's instructions as well as those described earlier14, 15, 16. Protein content was determined by using a BCA protein assay kit (Beytime Institute of Biotechnology, China).
Colony-forming assay
The division of cell groups was as described above. Cells were cultured in plastic flasks; 48 h after transfection, the flasks were irradiated with indicated dose of X-rays (0, 2, 4, 6, 8, 10, 15 and 20 Gy). After IR, cells were immediately trypsinized, diluted, counted and seeded in dishes with a diameter of 60 mm at various cell densities. Three replicates were set at each radiation dose of each group. After 2 weeks of incubation, cell colonies were stained with crystal violet dissolved in methanol. The colonies containing more than 50 cells were counted. The plating efficiency (PE) of each group was calculated by PE = (colony number/inoculating cell number)×100%. The survival fraction of each group was corrected by SF=PE (irradiated group)/PE (unirradiated group) × 100%. A dose-survival curve was obtained for each experiment and used for calculating the radiobiological parameters. The cell-survival curve was fitted using SPSS 13.0 software according to the multi-target single-hit model. The equation of SF = 1-(1-e-D/D0)N was applied to calculate the cellular radiosensitivity (mean lethal dose, D0), the capacity for sublethal damage repair (quasithreshold dose, Dq), the dose for 37% survival (D37), the extrapolationnumber (N) and the survival fraction (SF) after irradiation at a dose of 10 Gy (SF10).
Measurement of intracellular ROS
The division of cell groups was the same to that mentioned above. Forty-eight hours after transfection, cells were irradiated with 10 Gy of X-rays, and intracellular ROS were detected using the cell-permeable probe DCFH-DA (Invitrogen) dissolved in high quality anhydrous dimethylsulfoxide (DMSO). Three replicates were set at each observation point of each group. The cells were treated with either DCFH-DA (final concentration of 5 μmol/L) or non-oxidizable control (final concentration of 1 μmol/L) probe at 0 h, 1 h, and 3 h after IR, and then incubated in 5% CO2 at 37 °C. After 30 min, the cells were collected by trypsinization and washed with cold phosphate buffered saline (PBS) 3 times. Then they were resuspended in cold PBS at a concentration of 1×106 cells/mL, and fluorescence was detected by flow cytometry.
Apoptosis analysis
The division of cell groups was as described above. Six replicates were set at each group. Cells were irradiated with 10 Gy of X-rays 48 h after transfection. The cells were collected by trypsinization 24 h post IR, washed twice with cold PBS, and then resuspended in 1×binding buffer at a concentration of 1×106 cells/mL. These mixtures (100 μL) were then transferred to 1.5 mL eppendorf tubes respectively and 5 μL of fluorescein-conjugated Annexin V (Annexin V-FITC) and 10 μL of propidium iodide (PI) (Pharmingen, San Diego, CA, USA) were added to each tube. The cells were gently vortexed and incubated for 15 min under room temperature in the dark. Then, 400 μL of binding buffer was added to each tube. Flow cytometry was performed within 1 h. Apoptosis was determined by the percentage of cells stained positively by Annexin V-FITC and PI. Multiparameter cytometric measurements were performed using the minimum of 10 000 events for each sample. The data were analyzed with LYSIS II software (Becton Dickinson, Buccinasco, Mi, Italy).
Tumor xenograft experiments
Eca-109 cells were chosen for in vivo test. Twenty-five nude mice were divided into 5 groups (5 in each group) as follows: Blank group (implanted with control Eca-109 cells), Prx1 siRNA group (implanted with control Eca-109 cells and later transfected with Prx1 siRNA), Prx5 shRNA group (implanted with Eca-109 cells stably expressing Prx5 shRNA), Combined group (implanted with Eca-109 cells stably expressing Prx5 shRNA and later transfected with Prx1 siRNA), and Combined Neg group (implanted with Eca-109 cells stably expressing a non-targeted control Prx5 shRNA and later transfected with Prx1 siNeg). The in vivo treatment was performed in the following steps. First, 2 × 105 of each type of Eca-109 cells (those stably expressing Prx5 shRNA, those stably expressing a non-targeted control shRNA, and control cells) were suspended in 200 μL of PBS and injected subcutaneously into the upper hind leg of each mouse in the corresponding group. Second, 7 days after implantation, the tumor reached 4−5 mm in diameter, and intratumoral injections were conducted: Prx1 siRNA (0.5 mg/kg) in the Prx1 siRNA group and Combined group, and Prx1 siNeg (0.5 mg/kg) in the Combined Neg group. The siRNA was mixed with 50 μL Lipofectamine 2000 and proper volume of Opti-MEM medium (Life Technologies, USA) to the final volume of 100 μL17. The same volumes of the mixture without siRNA were injected in the mice in Blank group and Prx5 shRNA group. Forty-eight hours following intratumoral injection, IR was performed in all the groups. Tumor sizes were measured by a caliper every three days and the volumes were estimated using the following formula: tumor volume = 0.5×(tumor length)×(tumor width)2. Thirty-one days later, all the mice were sacrificed.
Statistical analysis
Statistical analysis was performed with SPSS 13.0 for Windows (Scientific Packages for Social Sciences, Chicago, IL, USA) and the results were statistically evaluated by one-way analysis of variance. The difference was considered statistically significant when P<0.05.
Results
High expression of Prx1 and Prx5 in Eca-109 and TE-1 cells
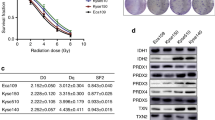
The mRNA expression profiles of Prxs in cell lines Eca-109 and TE-1 were analyzed by real-time PCR assay, calculating the relative gene expression by normalizing the results with β-actin mRNA expression. The β-actin normalized mRNA levels of six different Prx isoforms in both Eca-109 and TE-1 cells were shown in Figure 1A and 1B. Among the six Prx isoforms, Prx4 expression was the lowest. Prx1 and Prx5 expressions were significantly higher than Prx2, Prx3, Prx4 and Prx6 expressions in both cell lines (P<0.05, P<0.05). Results of representative immunodetection of Prxs (Prx1, Prx2 and Prx5) were in accordance with their mRNA expressions in Eca-109 and TE-1 cells (Figure 1C).

The expression profiles of Prxs in cell lines Eca-109 and TE-1. The mRNA expression levels of six different Prx isoforms were detected by real-time PCR assay; the mRNA levels for Prxs were normalized by the mRNA level of β-actin. (A) The mRNA expression profiles of Prxs in Eca-109. Prx1 and Prx5 are relatively highly expressed (bP<0.05 vs Prx2, Prx3, Prx4 and Prx6). (B) The mRNA expression profiles of Prxs in TE-1 cells. Prx1 and Prx5 are relatively highly expressed (bP<0.05 vs Prx2, Prx3, Prx4 and Prx6). (C) Representative Western blot analysis of Prxs (Prx1, Prx2 and Prx5) in Eca-109 and TE-1 cells. The results were in accordance with their mRNA expressions in Eca-109 and TE-1 cells.
Down-regulation of Prx1 and Prx5 by RNAi
Based on the profiles of Prx mRNA expression in Eca-109 and TE-1 cells, the protein expressions of Prx1 and Prx5 were silenced using siRNA and shRNA, respectively. Clones of Eca-109 and TE-1 cells stably expressing Prx5 shRNA or a non-targeted control shRNA were successfully established. The representative results of immunodetection of Prx1 and Prx5 in different cell groups of Eca-109 and TE-1 were shown in Figure 2. Protein loading equivalence of Prx1 and Prx5 was assessed by the protein expression of β-actin. It was shown that Prx1 and/or Prx5 were efficiently silenced by Prx1 siRNA and/or Prx5 shRNA in both Eca-109 and TE-1 cells.

The protein expressions of Prx1 and/or Prx5 in different cell groups 72 h post transfection. The protein expressions of Prx1 and/or Prx5 decreased in Prx1 siRNA group, Prx5 shRNA group and Combined group, compared with those in Blank group, Prx1 siNeg group, Prx5 shNeg group and Combined Neg group.
Radiosensitivity of Eca-109 and TE-1 cells with down-regulated Prx1 and/or Prx5
As mentioned in Materials and methods, Eca-109 and TE-1 cells were divided respectively into 7 groups, ie, altogether 14 cell groups. For detection of cell radiosensitivity, a total of 24 replicates were set for each group of cells. Forty-eight hours after transfection, the replicates of each cell group were irradiated with different doses (0, 2, 4, 6, 8, 10, 15 and 20 Gy), respectively. After 14-d incubation, clonogenic survival of each group was assessed. The cell-survival curve was fitted according to the multi-target single-hit model (Figure 3A, 3B). As the curves for the control groups are similar, we adopt only that of Blank group to represent the cell survival of all the 4 control groups. Relative biological parameters for different cell groups are shown in Table 2. It was found that D0, Dq and D37 of Prx1 siRNA group, Prx5 shRNA group and combined group in both Eca-109 and TE-1 cells were remarkably lower than those in control groups, which suggested that the cells were insensitive to low dose of radiation. While with 10 Gy of radiation, the survival fraction (SF) of the cells decreased obviously in all groups. As shown in Table 2, SF10 was significantly lower in Prx1 siRNA group, Prx5 shRNA group and Combined group as compared to the control groups (P<0.05).

Clonogenic survival of Eca-109 cells (A) and TE-1 cells (B) 14 d post IR. All the cell groups received 0–20 Gy of IR 48 h after transfection, and cloning formation was detected by 14 d after irradiation.
Influence of Prx1 and/or Prx5 down-regulation on the activities of other antioxidant enzymes
With Prx1 and/or Prx5 was down-regulated by RNAi, the activities of other antioxidant enzymes (CAT, GPx, Cu/Zn-SOD and Mn-SOD) were detected before IR by enzyme-linked immunosorbent assay, and the results are shown in Table 3. No remarkable change of the activities of the enzymes was found in the treated groups and the control groups (P>0.05).
Increase of tumor cell intracellular ROS by RNAi in vitro
The results showed that intracellular ROS increased significantly in all cell groups 1 h after 10 Gy of radiation, especially in Prx1 siRNA group, Prx5 shRNA group and Combined group (Figure 4A & 4B). No significant difference in ROS levels was observed among the control groups (so the data are not shown). Figure 4C shows that, compared with that in Blank group, the ROS levels were higher in the cells transfected with Prx1 siRNA and/or Prx5 shRNA (P<0.05) at the three observation points, namely, 0 h, 1 h and 3 h. The ROS levels reached the peak at 1 h, while decreased notably at 3 h.

Change of intracellular ROS post transfection and 10 Gy of IR in vitro. The levels of ROS were determined using the dye DCFH-DA. Flow cytometric histograms show a broad unimodal distribution of DCF fluorescence in different cell groups. Increase in the ROS levels is indicated as a shift in the histograms. (A) Increased intracellular ROS in Eca-109 cells. (B) Increased intracellular ROS in TE-1 cells. As shown in (A) and (B), ROS increase significantly in all cell groups 1 h post IR. The levels of ROS are higher in the cells transfected with Prx1 siRNA and/or Prx5 shRNA, P<0.05 vs Blank group. (C) The change of intracellular ROS levels at different observation points. The ROS levels are higher in the cells transfected with Prx1 siRNA and/or Prx5 shRNA at the three observation points (P<0.05 vs Blank group), particularly in the cells transfected with both Prx1 siRNA and Prx5 siRNA (P<0.05 vs Prx1 siRNA and Prx5 shRNA groups).
Increase of IR-induced apoptosis rates of Eca-109 and TE-1 cells by RNAi in vitro
IR-induced apoptosis in each group was analyzed by flow cytometry 24 h after 10 Gy of radiation (Figure 5A & 5B). The results showed an increase in IR-induced apoptosis rates when Prx1 and/or Prx5 were decreased by RNAi. Furthermore, in cells where both Prx1 and Prx5 were decreased (Combined group), apoptosis rate was higher than that in cells where Prx1 or Prx5 was decreased alone (P<0.05, P<0.05 and P<0.05). No significant difference in apoptosis rate was observed among control groups.

Change of tumor cell apotosis after transfection and 10 Gy of IR in vitro. The cells were exposed to 10 Gy of IR 48 h post transfection. IR-induced apoptosis in each group was analyzed by flow cytometry 24 h after IR. (A) Representative flow cytometry profiles of Eca-109 cell groups. It is shown that knockdown of Prx1 and/or Prx5 increased apoptosis. (B) Representative flow cytometry profiles of TE-1 cell groups. It is shown that knockdown of Prx1 and/or Prx5 increased apoptosis. All the data are means±SEM. n=3. bP<0.05 vs Blank and Combined groups; eP<0.05 vs Blank and Combined groups; hP<0.05 vs Blank, Prx1 siRNA, Prx5 shRNA, and Combined Neg groups.
Enhancement of tumor radiosensitivity by RNAi in vivo
A tumor xenograft model was used to examine the radiosensitizing effect of Prx1 siRNA and Prx5 shRNA in vivo. The tumors in mice grew to final volume between 273 and 1915 mm3 thirty-one days after implantation. The growth of tumor was delayed by RNAi and IR in Prx1 siRNA group (930.40±267.70 mm3), Prx5 shRNA group (680.20±240.55 mm3) and Combined group (364.00±90.77 mm3), compared to Blank group (1537.20 ± 347.90 mm3) and Combined negative group (1455.80 ± 294.91 mm3) (P<0.05, P<0.05 and P<0.05). It is found that the tumor growth in Combined group was more significantly inhibited than that in either Prx1 siRNA group or the Prx5 shRNA group (P<0.05, Figure 6).

Tumor growth post transfection and IR in vivo. The long black arrow indicates intratumoral injection of Prx1 siRNA in Prx1 siRNA group and Combined group, and Prx1 siNeg in Combined Neg group. The short black arrow indicates 10 Gy of IR 48 h after the injection. Each point on a curve represents the mean and standard deviation of tumor mass from the 5 mice at each measuring point. It can be seen that the final tumor growth was delayed in Prx1 siRNA group (bP<0.05 vs Blank, Prx5 shRNA, and Combined groups), Prx5 shRNA group (eP<0.05 vs Blank, Prx1 siRNA, and Combined groups) and Combined group, (hP<0.05 vs Blank, Combined Neg, Prx1 siRNA, and Prx5 shRNA groups).
Discussion
We have found in this study that only two Prx isoforms (Prx1 and Prx5) are highly expressed in Eca-109 and TE-1 cells, and silencing Prx1 and/or Prx5 sensitizes esophageal cancer cells to IR through accumulation of intracellular ROS. This is the report on the Prx expression profiles in esophageal cancer cells and the influence of Prx1 and Prx5 expressions on the radiosensitivity of esophageal cancer cells.
ROS are usually a side product of general metabolism. Intracellular ROS can be induced by IR and serve as major mediators of radiation damage18. When ROS generation exceeds the cellular antioxidant defenses, cell damage ensues19, 20. In the present study, we find that Prx1 and/or Prx5 knock-down through RNAi, together with the IR treatment after the transfection, lead to significant elevation of the levels of intracellular ROS and the cell apoptosis rates. Thus, increased intracellular ROS could be, in part, due to the lower Prx1 and/or Prx5 levels in tumor cells. This assumption is also supported by some studies21, 22, 23. Furthermore, the more Prx isoforms that are silenced, the more intracellular ROS remained unscavenged in the tumor cells and the more tumor cells die post IR. After IR, the dose-survival curves of Eca-109 or TE-1 cells in Prx1 siRNA and/or Prx5 shRNA groups exhibit a narrower shoulder and a greater slope rate, indicating decreased quasi-threshold dose (Dq) and mean lethal dose (D0), respectively, that suggests that the radiosensitivity of the cells has increased. In our further investigation of the potential radiosensitizing effects of Prx1 siRNA and Prx5 shRNA in vivo, we have achieved similar results. So, our findings show that down-regulation of Prx1 and Prx5 can enhance the radiosensitivity of Eca-109 and TE-1 cells.
The enzymatic activity of other primary antioxidant enzymes (CAT, GPx, Cu/Zn-SOD, and Mn-SOD) in the cancer cells has not changed when Prx1 and/or Prx5 are inhibited. In another word, decreasing Prx1 and/or Prx5 does not alter the activities of the enzymes. This suggests that other antioxidant enzymes will not compensate for the loss of Prx1 and/or Prx5. All our findings indicate that tumor cell radiosensitivity is influenced by altered levels of Prxs, and Prx1 and Prx5 play a critical role in regulating intracellular ROS.
In addition to the protective role of Prx1 and Prx5 as peroxidase, evidence has indicated other mechanisms of them involved in protecting cells from radiation-induced death. It is reported that Prx1 plays a negative role in apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis. ASK1 is a mitogen-activated protein kinase (MAPK) kinase kinase which phosphorylates c-Jun N-terminal kinase (c-JNK) and p38 MAPK, and elicits apoptotic response24. ASK1 activity is regulated in multiple ways, one of which is through interaction with Prx1. The overexpression of Prx1 inhibits the activation of ASK1, and results in the inhibition of downstream signaling cascades such as the mitogen-activated protein kinase kinase 3/6 (MKK3/6), p38 and c-JNK pathway25. Moreover, overexpression of Prx1 could directly suppress IR–induced c-JNK activation and cell apoptosis through interaction with the glutathione S-transferase π (GSTπ)-JNK complex26. While, in Prx1 knock-down cells, ASK1, p38, and c-JNK are quickly activated, leading to cell apoptosis in response to H2O225. The cell cycle phase also determines a cell's relative radiosensitivity, with cells being most radiosensitive in the G2-M phase, less sensitive in the G1 phase, and least sensitive during the latter part of the S phase27. It has been shown that Prx1 expression is associated with cell cycle phase distribution. After IR, cell cycle phases are redistributed to radiosensitive phases in Prx1 knock-down cells, that is, G2-M checkpoint arrested, more cells blocked in G2 phase, and higher ratio of G2/G1 observed21. In the presence of excess peroxide, the cysteine residue of Prxs is found to be hyperoxidized to the sulfinic acid or to the sulfonic acid to form complex oligomeric structures28, this hyperoxidized Prx toroids are reported to exhibit a chaperone-like function protecting mammalian cells from hydrogen peroxide-induced apoptosis29, 30. A recent study demonstrated that the hyperoxidation of the active-site cysteine of Prx to cysteine sulfonic acid enhanced its molecular chaperone activity31.
Prx5 has been proved to have a broad range of functions that the expression of the redox-negative Prx5 mainly affects the mitochondrial pathway of apoptosis, and the impairment of the Prx5 enzymatic function also affects transmembrane potential and calcium loading capacity of mitochondria, as well as mitochondrial morphology32. For as yet unknown reasons, Prx5 is insensitive to hyperoxidation. To date, there has been no investigation of the chaperone activity of the Prx5. It is expected that these functions of Prx1 and Prx5 do not necessarily depend on peroxidase activity. Additionally, decreasing the expression of Prx1 and/or Prx5 in Eca-109 and TE-1 cells could also abrogate such functions, and, in turn, contribute to increased radiosensitivity.
Conclusion
Silencing Prx1 and/or Prx5 by RNAi can sensitize human esophageal cancer cells Eca-109 and TE-1 cells to IR, and the resulting intracellular ROS accumulation may contribute to the radiosensitizing effect of the RNAi. The results of this study suggest that inactivation of Prx1 and Prx5 may be a promising approach to improving the radiotherapy outcome of esophageal cancer.
Author contribution
Prof Jun ZHANG designed the research and revised the manuscript; Mai-cang GAO, Xiao-di JIA and Qi-fei WU conducted the research, analyzed the data and wrote the paper; Yan CHENG and Fen-rong CHEN helped with part of the research.
References
Ekman S, Dreilich M, Lennartsson J, Wallner B, Brattström D, Sundbom M, et al. Esophageal cancer: current and emerging therapy modalities. Expert Rev Anticancer Ther 2008; 8: 1433–48.
Herskovic A, Martz K, al-Sarraf M, Leichman L, Brindle J, Vaitkevicius V, et al. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med 1992; 326: 1593–8.
Gebski V, Burmeister B, Smithers BM, Foo K, Zalcberg J, Simes J . Survival benefits from neoadjuvant chemoradiotherapy or chemotherapy in esophageal carcinoma: a meta-analysis. Lancet Oncol 2007; 8: 226–34.
Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB . Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res 2001; 61: 3894–901.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009; 458: 780–3.
Kinnula VL, Lehtonen S, Sormunen R, Kaarteenaho-Wiik R, Kang SW, Rhee SG, et al. Overexpression of peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. J Pathol 2002; 196: 316–23.
Kang SW, Rhee SG, Chang TS, Jeong W, Choi MH . 2-Cys peroxiredoxin function in intracellular signal transduction: therapeutic implications. Trends Mol Med 2005; 11: 571–8.
Karihtala P, Mäntyniemi A, Kang SW, Kinnula VL, Soini Y . Peroxiredoxins in breast carcinoma. Clin Cancer Res 2003; 9: 3418–24.
Qi Y, Chiu JF, Wang L, Kwong DL, He QY . Comparative proteomic analysis of esophageal squamous cell carcinoma. Proteomics 2005; 5: 2960–71.
Ai J, Tan Y, Ying W, Hong Y, Liu S, Wu M, et al. Proteome analysis of hepatocellular carcinoma by laser capture microdissection. Proteomics 2006; 6: 538–46.
Cheng Y, Zhang J, Li Y, Wang Y, Gong J . Proteome analysis of human gastric cardia adenocarcinoma by laser capture microdissection. BMC Cancer 2007; 7: 191.
Zhang B, Wang Y, Su YP . Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett 2009; 286: 154–60.
Thomas S, Muralidharan A, Shah GV . Knock-down of calcitonin receptor expression induces apoptosis and growth arrest of prostate cancer cells. Int J Oncol 2007; 31: 1425–37.
He Z, Sun X, Mei G, Yu S, Li N . Nonclassical secretion of human catalase on the surface of CHO cells is more efficient than classical secretion. Cell Biol Int 2008; 32: 367–73.
An R, Dong C, Lei Y, Han L, Li P, Chen J, et al. PrP mutants with different numbers of octarepeat sequences are more susceptible to the oxidative stress. Sci China C Life Sci 2008; 51: 630–9.
Siafakas AR, Wright LC, Sorrell TC, Djordjevic JT . Lipid rafts in Cryptococcus neoformans concentrate the virulence determinants phospholipase B1 and Cu/Zn superoxide dismutase. Eukaryot Cell 2006; 5: 488–98.
Liu TG, Yin JQ, Shang BY, Min Z, He HW, Jiang JM, et al. Silencing of hdm2 oncogene by siRNA inhibits p53-dependent human breast cancer. Cancer Gene Ther 2004; 11: 748–56.
Mikkelsen RB, Wardman P . Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003; 22: 5734–54.
Gamaley IA, Klyubin IV . Roles of reactive oxygen species: Signaling and regulation of cellular functions. Int Rev Cytol 1999; 188: 203–55.
Cerutti PA . Prooxidant states and tumor promotion. Science 1985; 227: 375–81.
Zhang B, Su Y, Ai G, Wang Y, Wang T, Wang F . Involvement of peroxiredoxin I in protecting cells from radiation-induced death. J Radiat Res 2005; 46: 305–12.
Chen WC, McBride WH, Iwamoto KS, Barber CL, Wang CC, Oh YT, et al. Induction of radioprotective peroxiredoxin-I by ionizing irradiation. J Neurosci Res 2002; 70: 794–8.
Chen MF, Lee KD, Yeh CH, Chen WC, Huang WS, Chin CC, et al. Role of peroxiredoxin I in rectal cancer and related to p53 status. Int J Radiat Oncol Biol Phys 2010; 78: 868–78.
Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001; 2: 222–8.
Kim SY, Kim TJ, Lee KY . A novel function of peroxiredoxin 1 (Prx-1) in apoptosis signal-regulating kinase 1 (ASK1)-mediated signaling pathway. FEBS Lett 2008; 582: 1913–8.
Kim YJ, Lee WS, Ip C, Chae HZ, Park EM, Park YM . Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res 2006; 66: 7136–42.
Pawlik TM, Keyomarsi K . Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys 2004; 59: 928–42.
Seo JH, Lim JC, Lee DY, Kim KS, Piszczek G, Nam HW, et al. Novel protective mechanism against irreversible hyperoxidation of peroxiredoxin: Nα-terminal acetylation of human peroxiredoxin II. J Biol Chem 2009; 284: 13455–65.
Moon JC, Hah YS, Kim WY, Jung BG, Jang HH, Lee JR, et al. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J Biol Chem 2005; 280: 28775–84.
Lee W, Choi KS, Riddell J, Ip C, Ghosh D, Park JH, et al. Human peroxiredoxin 1 and 2 are not duplicate proteins: the unique presence of Cys83 in Prx1 underscores the structural and functional differences between Prx1 and Prx2. J Biol Chem 2007; 282: 22011–22.
Lim JC, Choi HI, Park YS, Nam HW, Woo HA, Kwon KS, et al. Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J Biol Chem 2008; 283: 28873–80.
Kropotov A, Gogvadze V, Shupliakov O, Tomilin N, Serikov VB, Tomilin NV, et al. Peroxiredoxin V is essential for protection against apoptosis in human lung carcinoma cells. Exp Cell Res 2006; 312: 2806–15.
Acknowledgements
This study was supported by a grant from Clinical Disciplines' Key Project of Ministry of Health of China (No 2007-353).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gao, Mc., Jia, Xd., Wu, Qf. et al. Silencing Prx1 and/or Prx5 sensitizes human esophageal cancer cells to ionizing radiation and increases apoptosis via intracellular ROS accumulation. Acta Pharmacol Sin 32, 528–536 (2011). https://doi.org/10.1038/aps.2010.235
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2010.235
Keywords
This article is cited by
-
Pathological effects of ionizing radiation: endothelial activation and dysfunction
Cellular and Molecular Life Sciences (2019)
-
Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress
British Journal of Cancer (2013)
-
Isoliquiritigenin Enhances Radiosensitivity of HepG2 Cells via Disturbance of Redox Status
Cell Biochemistry and Biophysics (2013)
