
Abstract
Nicotinic acetylcholine receptors (nAChRs) are expressed throughout the hippocampus, and nicotinic signaling plays an important role in neuronal function. In the context of learning and memory related behaviors associated with hippocampal function, a potentially significant feature of nAChR activity is the impact it has on synaptic plasticity. Synaptic plasticity in hippocampal neurons has long been considered a contributing cellular mechanism of learning and memory. These same kinds of cellular mechanisms are a factor in the development of nicotine addiction. Nicotinic signaling has been demonstrated by in vitro studies to affect synaptic plasticity in hippocampal neurons via multiple steps, and the signaling has also been shown to evoke synaptic plasticity in vivo. This review focuses on the nAChRs subtypes that contribute to hippocampal synaptic plasticity at the cellular and circuit level. It also considers nicotinic influences over long-term changes in the hippocampus that may contribute to addiction.
Similar content being viewed by others


Introduction
The hippocampus has been widely studied as an important neuroanatomical structure during learning and memory. Nicotinic acetylcholine receptors (nAChRs) are expressed in many hippocampal cell types, and pharmacological studies have implicated nAChR signaling in hippocampus-related behaviors including attention, learning and memory1, 2, 3, 4. In addition to the role that endogenous nicotinic cholinergic signaling plays in hippocampal function, the addictive drug nicotine has been shown to have profound and long-lasting effects on hippocampal synaptic activity. Here we will describe the types of nAChRs expressed in the hippocampus, what is known about their basic function, and how signaling via nAChRs affects hippocampal synaptic plasticity. We will also discuss how nicotine addiction is facilitated by synaptic plasticity in the hippocampus.
Neuronal nicotinic acetylcholine receptors
Nicotinic acetylcholine receptors are ligand-gated ion channels formed from the combination of five individual subunits surrounding an aqueous pore. There have been a total of 17 distinct genes identified in vertebrates that encode nAChR subunits. The various nAChR subunits can form functional channels from heteromeric combinations, or in some cases from homomeric assemblies, such as the α7 subunit. The relatively large number of identified genes results in a wide array of possible subunit combinations, many of which have been demonstrated to form functional channels in heterologous expression systems5, 6. The α1, β1, γ, δ, and ɛ subunits are found in vertebrate muscle, while α2−α10, and β2−β4 are commonly referred to as the neuronal subunits. In mammalian brain, functional combinations of nAChR subunits primarily consist of heteromeric combinations of alpha and beta subunits, although the α7 subunit forms functional homomeric channels that are widely distributed in the mammalian CNS, including the hippocampus. Because of the large number of possible subunit combinations and the lack of absolute certainty in determining the specific subunits that are present in native nAChRs, it is common to refer to various receptors as “containing” particular nAChR subunits. These receptors are denoted with an asterisk indicating that a specific subunit is present (eg, β2* or α7* nAChRs)6. It should also be noted that the term “neuronal” in reference to nAChR subunits is less than perfect because some of these subunits also have been found in non-neuronal tissues7.
Cholinergic projections to the hippocampus
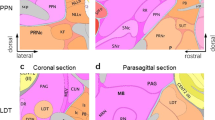
There is widespread cholinergic innervation throughout the brain to a variety of targets, including the hippo-campus8, 9. Generally speaking, these projections are diffuse and suggest broad, modulatory roles for cholinergic signaling at either muscarinic or nicotinic targets8, 10, 11. The medial septum-diagonal band complex is the primary source of cholinergic inputs to the hippocampus via the fimbria-fornix (Figure 1) 8, 9, 12, 13. The importance of these projections in maintaining normal cognitive function is highlighted by the fact that degeneration of the cholinergic neurons of the basal forebrain accompany cognitive deficits that arise is such degenerative pathologies as Alzheimer's disease14.

The major cholinergic afferent projections to the hippocampus. The hippocampus receives cholinergic innervation mainly from the medial septum-diagonal band complex via the fimbria-fornix. A fine network of cholinergic fibers projects to the hippocampus and dentate gyrus, and synaptic contacts are made onto pyramidal cells, granule cells, interneurons, and neurons of the hilus89. In addition to direct synaptic connections, ACh may spill out of synaptic contacts and also produce volume transmission within the hippocampus via non-synaptic signaling11, 50 (MS: medial septum, n Bas: nucleus Basalis, vDB: vertical Diagonal Band, hDB: horizontal Diagonal Band, SI: sublenticular substantia innominata, mHb: medial Habenula, LDT: Laterodorsal Tegmentum, PPT: Pedunculopontine Tegmentum, IPn: Interpeduncular nucleus. Adapted from© 1991, with permission from Elsevier 9).
nAChR subunit expression in the hippocampus
Neuronal nAChR subunits are known to be highly expressed throughout the hippocampus15, 16, 17. Early radioligand binding studies in the rodent brain showed robust labeling by both nicotine and α-bungarotoxin (α-btx, a selective antagonist of the α7 nAChR)18. Later confirmation of similar binding sites in human hippocampus followed19. In the rodent hippocampus, in situ hybridization for subunit-specific mRNA indicates that the α7 and β2 subunits have the highest expression15, 20, 21, 22, 23, 24, 25, 26, but many other nAChR subunit transcripts are also present. Similarly, the human hippocampus also shows widespread nAChR expression, particularly within the interneurons, but the majority of pyramidal neurons and dentate granule cells also are immunoreactive for multiple nAChR subunits27, 28.
The details of the normal physiological roles of nAChRs are still being elucidated, but there have been significant anatomical, pharmacological, and physiological advances. Thus far, hippocampal nAChRs have been identified as being expressed at presynaptic and preterminal sites, as well as being found at somatic and dendritic sites5, 26, 29, 30, 31, 32, 33. Exactly where the various nAChR subtypes are positioned, whether they are expressed alone or with other subtypes, and whether receptor activation or desensitization predominates determines how neuronal excitability and circuit activity are regulated (Figure 2). Furthermore, the subunit composition of the various nAChRs dictates their pharmacology and determines the magnitude and time course nAChR-mediated changes in membrane potential or increases in intracellular [Ca2+]3, 5, 6.

Schematic illustration showing some of the known subcellular regions of nAChR expression. Presynaptic and somatic expression of nAChRs, as well as their specific subunit composition can have important ramifications for how they affect neuronal function and plasticity. The combination of presynaptic facilitation of neurotransmitter release and postsynaptic depolarization and Ca2+ influx can enhance or possibly evoke synaptic plasticity (purple shows preterminal expression, light blue is presynaptic, and dark blue is postsynaptic or extrasynaptic. Adapted from© 2004 by Cold Spring Harbor Laboratory Press59.
Presynaptic neuronal nAChRs regulate the release of multiple neurotransmitters in the hippocampus
One of the best characterized roles for nicotinic receptors comes from studies of their presynaptic expression and regulation of neurotransmitter release34, 35. This general function of nAChRs has been shown to be important in a multitude of brain regions including (but not limited to) the ventral midbrain, prefrontal cortex, cerebellum, interpeduncular nucleus, and striatum36. In the hippocampus, activation of nAChRs has been shown to facilitate the release of several neurotransmitters including glutamate, GABA, and norepinephrine30, 34, 37, 38, 39, 40, 41, 42. The mechanisms of facilitation involve nAChR mediated increases in presynaptic calcium concentration, which can be accomplished either directly via the highly calcium-permeable α7* nAChRs, or through depolarization and subsequent activation of voltage-gated calcium channels16, 34, 39, 43, 44, 45, 46. In addition, the initial calcium influx is capable of producing further Ca2+ release from intracellular stores47. Presynaptic nAChR-mediated increases in glutamate release at hippocampal CA1 synapses is also known to contribute to the development of synaptic plasticity41, 42, a phenomenon that will be discussed in greater detail later in this review.
Somatic and dendritic nAChR expression
Unlike the classical fast synaptic transmission that typifies nAChR signaling at the neuromuscular junction and at peripheral nervous system ganglia, only a small minority of fast currents evoked by synaptically released acetylcholine have been reported in the hippocampus48. In spite of this, there is evidence for high levels of somatic nAChR expression. Patch-clamp studies using rodent brain slice preparations and non-synaptic agonist application have shown that β2* and α7* nAChRs are important mediators of nicotinic responses in the hippocampus. Mice lacking the α7 subunit lack the fast agonist-evoked currents that are characteristic of the α7* nAChRs49. Given the extent and variety of nAChR expression in this brain region, it is possible ACh that diffuses away from release sites can mediate nAChR currents that are presently underestimated11, 50. The diffuse nature of cholinergic innervation throughout the brain makes the isolation and experimental stimulation of well-defined groups of cholinergic fibers very difficult. The commonly used technique of brain slice electrophysiology severs a large number of axonal inputs, potentially reducing the activity of spontaneous cholinergic synaptic activity. There is also evidence for diffuse “en passant” synaptic release of ACh that may travel to non-synaptic sites, suggesting a role for volume transmission in hippocampal cholinergic signaling11, 50. The fact that the ACh metabolite choline is a selective agonist of α7* receptors51, 52 and that choline can desensitize these channels53 further supports volume transmission as a potentially important type of cholinergic signal in the hippocampus.
Nicotinic currents can be found in the majority of hippocampal neurons including principal cells. However, the greatest degree of nAChR expression is found in GABAergic interneurons. Again, α7* nAChR currents are major contributors in these cells, but there are also significant contributions of β2* nAChRs31, 32, 54. The activation of nAChRs on GABAergic interneurons can have important circuit level effects by either inhibiting or disinhibiting pyramidal neurons54, 55. Since there is evidence for stimulation of hippocampal GABAergic neurons by endogenous nicotinic cholinergic activity10, 48, 56, it is likely that low concentrations of nicotinic agonists could desensitize nAChRs and decrease the tonic inhibition produced by these inhibitory interneurons. A similar nAChR-mediated reduction in inhibition has been observed in vitro in the ventral midbrain57, 58, 59, 60. This type of reduction in tonic inhibition is especially likely in the case of exposure to nicotine, since the GABAergic interneurons express greater levels of β2* nAChRs, and this nAChR subtype has a high affinity for nicotine resulting in greater desensitization61, 62. Thus, nicotinic cholinergic transmission can regulate the firing frequency of hippocampal principal cells directly, and also through more complex modulation of GABAergic inhibition.
Nicotinic modulation of learning and memory behavior
Nicotinic agonists have long been known to improve certain types of memory-related behavior3, 4, 63. In animal studies, both acute and chronic nicotine administration improves working memory, and nicotinic agonists improve learning and memory in both humans and non-human primates64. By contrast, blockade of nicotinic function impairs memory. Selective infusion of nicotinic antagonists into either the basolateral amygdala, or the hippocampus has been shown to impair the working memory of rats in a radial arm maze task65, 66, 67, 68. In addition to these observations, some cognitive symptoms of Alzheimer's disease are improved by the clinical use of acetycholinesterase inhibitors. However, increased levels of acetylcholine do not selectively affect nAChRs, and there is evidence for the involvement of both nicotinic and muscarinic receptor function in learning and memory mechanisms63.
It is important to note that nAChR function seems to affect only specific forms of memory and that there are some discrepancies in the literature that suggest that experimental conditions are important for uncovering nicotinic effects on memory. For example, radial arm maze studies using rodents indicate that nicotinic agonists enhance working memory, but not reference memory64, 68. Studies using either pharmacology or selective cholinergic lesions suggest that cognitive impairment or the use of difficult behavioral tasks are necessary to reveal the memory benefits of enhanced nicotinic function69. In keeping with this general idea, nicotinic agonists have also demonstrated the ability to improve memory in aged animals70, 71. However, another study showed no improvement in working memory performance with aged rats given chronic nicotine70. Differences in experimental conditions and variations in the level of basal cholinergic function between species or strains of animals may contribute to these discrepancies. The apparent sensitivity of these effects of nicotinic function on memory further supports the idea of a primarily modulatory role in this type of behavior. That is, if cholinergic functioning is normal or intact, then experimental manipulation of nicotinic function may not produce observable memory improvements. But, if the subject is impaired or presented with a difficult task, then nicotinic activity can serve to improve or normalize performance.
In addition to this well documented, but complex role for nAChR function in learning and memory behavior, in vitro experiments have also shown a role for nicotinic receptor function in hippocampal network activity. Specifically, the α7* nAChR has been shown to be involved in the induction of gamma (30−80 Hz) oscillations in rat brain hippocampal slices72, a firing pattern linked to attention, learning, and memory.
Synaptic plasticity underlies learning and memory and is modulated by nAChRs
For several decades the cellular processes of synaptic plasticity have been hypothesized to subserve learning and memory behavior73. There are multiple forms of synaptic plasticity that have been demonstrated in the hippocampus, including short-term potentiation (STP), long-term depression (LTD), and long-term potentiation (LTP), all of which have been associated with memory74. LTP, a long lasting activity-dependent strengthening of synaptic transmission, came to represent the physiological fulfillment of Hebb's postulate regarding coincident activity of neurons75. This hypothesis was put forth many years ago, and the evidence supporting the hypothesis has culminated in direct support for the relationship between hippocampal LTP and memory measured in freely moving mice76. LTP can be induced experimentally in a variety of ways, but a common method of in vitro LTP induction involves the pairing of a presynaptic stimulation with a postsynaptic depolarization. In the case of nicotinic receptors, the combination of nAChR mediated presynaptic facilitation of excitatory neurotransmitter release with a coincident postsynaptic stimulus can either directly produce synaptic plasticity, or modulate its direction or duration41, 42, 77, 78. This dual mechanism of presynaptic facilitation of glutamate release coupled with postsynaptic depolarization and subsequent increases in intracellular [Ca2+] allows nAChRs to function to produce or enhance the kind of coincidence that is necessary for synaptic plasticity to occur.
The timing and location of nAChR activation regulates hippocampal synaptic plasticity
The type of nAChR-mediated modulation of synaptic plasticity that is described in the previous section is known to be temporally and spatially restricted. As previously mentioned, activation and desensitization of nAChRs expressed by the inhibitory GABAergic interneurons in the hippocampus can have a significant net effect on the activity and output of principal cells. An example of this was seen in experiments where LTP was induced by high frequency stimulation at Schaffer collateral-CA1 synapses41. By varying the location of an ACh application pipette, it was possible to apply ACh in a manner that changed LTP to STP (in this case potentiation lasting ≤20 min). This occurs owing to the activation of nAChRs on neighboring GABAergic interneurons, which are known to directly inhibit CA1 pyramidal neurons41, 54, 79. A similar effect can be produced with GABAergic neurons having inhibitory connections to other inhibitory neurons. In that case, activation of inhibitory neurons leads to inhibition of GABA neurons that inhibit pyramidal neurons. Thus, there is overall disinhibition of the excitatory circuit, producing a net increase in principal cell activity through disinhibition54.
The timing of nAChR activity also can modulate synaptic plasticity. Properly timed nAChR activity can add further excitation that shifts hippocampal STP to LTP42. In mouse brain slices, STP was induced by pairing weak stimulation of Schaffer collateral-CA1 synapses with a depolarization of the postsynaptic CA1 pyramidal neuron (Figure 3A). The placement of a puffer pipette containing ACh in the dendritic field of the pyramidal cell showed that brief agonist applications alone (sufficient to drive action potentials in the postsynaptic neuron) were not sufficient to induce synaptic plasticity. However, if ACh was applied 2 s before the STP stimulus induction paradigm, a potentiation lasting at least one hour developed (Figure 3B). In other words, the brief 2 s pre-application of ACh boosted the induction of STP to LTP. This effect required ACh since action potentials evoked by a depolarizing current injection in the postsynaptic cell did not produce a similar increase in the duration of potentiation. A closer examination of the timing of ACh delivery revealed that if the last ACh-induced action potential preceded electrical stimulation by more than 5 s or occurred after the stimulation, then there was no enhancement of synaptic potentiation. Only if the last ACh-induced action potential occurred prior to stimulation within a range of 1–5 s, then LTP was produced. Interestingly, if the last ACh-induced action potential occurred <1 s prior to stimulation, then LTD rather than LTP occurred. Since these time-dependent differences in the sign and duration of plasticity occur over the course of seconds rather than milliseconds, there is a relatively broad range in which either endogenous ACh or exogenous nicotine may alter the nature of hippocampal excitatory outputs. It seems likely that the exact timings delineated in the in vitro slice system will be different than those present in vivo, but the results indicate that the timing of nAChR activity can dictate the plasticity consequences of afferent activity.

Nicotinic activation can shift STP to LTP within a specific time window. (A) Schematic illustration showing the placement of the stimulating electrode in the Schaffer collateral pathway, the recording electrode on the CA1 pyramidal cell, and the ACh application pipette in the dendrites of the CA1 neuron. For this experiment, stimulation was given at 100 Hz for 1 s and depolarization of the postsynaptic cell was induced by a 100 pA current injection for 1 s. The ACh concentration in the puffer pipette was 1 mmol/L and drug applications were from 0.5–1 s in duration. (B) Recordings of postsynaptic potentials in a CA1 pyramidal cell. This recording shows the LTP that occurs when ACh is puffed 2 s prior to the delivery of the stimulation/depolarization paradigm that normally produces STP. Adapted from © 2005 by the Society for Neuroscience42.
Nicotinic signaling regulates hippocampal synaptic plasticity in vivo
In addition to the in vitro evidence for nicotinic contributions to synaptic plasticity, there are studies showing similar findings in vivo. In anesthetized mice, nicotine has been shown to produce LTP at perforant path synapses in the dentate gyrus in the absence of afferent stimulation80. This differs from the in vitro situation where nicotine modulates or enhances existing synaptic plasticity brought on by high frequency stimulation. Since this was observed using systemic delivery of relatively high concentrations of nicotine in intact, anesthetized animals, it is difficult to make direct comparisons to the in vitro data. However, there are similarities in the time course and magnitude of the effect80. This same study showed that both nicotine-evoked LTP, and the LTP induced by high frequency stimulation are blocked by pretreatment with the non-selective nAChR antagonist, mecamylamine, suggesting that stimulation induced LTP requires endogenous cholinergic signaling via nAChRs. Furthermore, the β2* selective agonist epibatidine, as well as the α7* nAChR selective agonist choline were also able to produce a dose-dependent, in vivo LTP80.
A recent study using transgenic mice showed that chronic nicotine upregulated the expression of the α4 nAChR subunit in the glutamatergic afferents of the medial perforant path from the entorhinal cortex to the dentate gyrus in the hippocampus81. Mice treated with a similar chronic nicotine paradigm had hippocampal brain slices that showed an enhancement of LTP when given acute nicotine with tetanic stimulation of the medial perforant path. Interestingly, this sensitization of excitatory inputs to the dentate gyrus was paralleled by a similar upregulation of α4 nAChR subunits in the inhibitory GABAergic neurons of the ventral midbrain, resulting in a concomitant decrease in nicotine-mediated dopaminergic activity, possibly contributing to nicotine tolerance81.
Hippocampal synaptic plasticity and nicotine addiction
One of the more challenging public health problems faced by society directly relates to the long-lasting changes in synaptic function that are brought on by the addictive drug, nicotine. There is a growing trend in the field of addiction research to view the problem, in part, as a disorder that involves learning and memory82. This hypothesis is supported by evidence that several addictive drugs, including nicotine, produce LTP in the reward-linked dopaminergic neurons of the ventral midbrain83. The hippocampus is recruited in this process as environmental and contextual cues are given salience by drug-enhanced activity of dopaminergic afferents. It is commonly reported in the epidemiological literature that specific environments will produce drug craving in humans, particularly those associated with previous drug use84. Furthermore, it has been shown in animal models that environmental cues can reinstate previously extinguished drug seeking behaviors84. Nicotine is an addictive drug that has a variety of profound, long-lasting, and direct effects on synaptic function in the hippocampus that may synergize with its effects on the ventral midbrain reward circuitry.
A large number of people continue to initiate tobacco use despite widespread efforts to educate the public regarding its addictive nature and potential health hazards85. Once the addiction process has progressed, tobacco users often report the desire to quit and frequently make several attempts with varying degrees of success and a very high rate of relapse85. In addition to psychosocial therapy, there are pharmacological treatments that have been approved for use in humans. These include nicotine replacement therapy, bupropion (Zyban®), and varenicline (Chantix®). Nicotine replacement therapy aims to use either oral nicotine (in the form of chewing gum or lozenges) or transdermal nicotine (a sustained release patch) to provide enough nicotine to lessen withdrawal symptoms, while preventing exposure to the known carcinogens and other hazardous substances in tobacco. Bupropion has been reported to be a nicotinic antagonist at relatively high concentrations86, but at clinically relevant doses, its primary mechanism of action is as a weak inhibitor of catecholamine reuptake86. It is interesting to note that bupropion is also prescribed for the treatment of depression under a different trade name (Wellbutrin®), suggesting that its utility as a smoking cessation aid may be related to its ability to mitigate some of the more aversive subjective experience associated with nicotine withdrawal. Varenicline is a relatively recently approved drug and was originally characterized as a partial agonist of α4β2* nAChRs87, but it has also been reported to be an agonist of α7* nAChRs88. It has been hypothesized that as a partial agonist of α4β2* nAChRs, varenicline provides an optimal, sub-maximal activation of the high-affinity nicotine receptor, while at the same time, competitively inhibiting nicotine itself from binding87.
For each of these pharmacological approaches to smoking cessation therapy, it is currently unclear how hippocampal synaptic plasticity is affected, primarily because the relationship has not been studied in great detail. However, it is interesting to speculate that specific targeting of synaptic plasticity in various relevant brain regions may aid in the treatment of nicotine addiction. For example, future drug therapies may attempt to separate the strong relationships between certain environmental cues and the subjective experience of drug craving by working to counteract nicotine-induced synaptic plasticity selectively. Of course, any sort of drug design that would use this type of therapeutic strategy would require a better understanding of the basic mechanisms involved in nicotine addiction.
References
Alkondon M, Albuquerque EX . The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog Brain Res 2004; 145: 109–20.
Liu Z, Zhang J, Berg DK . Role of endogenous nicotinic signaling in guiding neuronal development. Biochem Pharmacol 2007; 74: 1112–9.
Dani JA, Bertrand D . Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol 2007; 47: 699–729.
Kenney JW, Gould TJ . Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol Neurobiol 2008; 38: 101–21.
Albuquerque EX, Pereira EF, Alkondon M, Rogers SW . Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 2009; 89: 73–120.
Millar NS, Gotti C . Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009; 56: 237–46.
Sharma G, Vijayaraghavan S . Nicotinic receptor signaling in nonexcitable cells. J Neurobiol 2002; 53: 524–34.
Kasa P . The cholinergic systems in brain and spinal cord. Prog Neurobiol 1986; 26: 211–72.
Woolf NJ . Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol 1991; 37: 475–524.
Hefft S, Hulo S, Bertrand D, Muller D . Synaptic transmission at nicotinic acetylcholine receptors in rat hippocampal organotypic cultures and slices. J Physiol 1999; 515: 769–76.
Descarries L, Gisiger V, Steriade M . Diffuse transmission by acetylcholine in the CNS. Prog Neurobiol 1997; 53: 603–25.
Alonso JR, Amaral DG . Cholinergic innervation of the primate hippocampal formation. I. Distribution of choline acetyltransferase immunoreactivity in the Macaca fascicularis and Macaca mulatta monkeys. J Compar Neurol 1995; 355: 135–70.
Yoshida K, Oka H . Topographical projections from the medial septum-diagonal band complex to the hippocampus: a retrograde tracing study with multiple fluorescent dyes in rats. Neurosci Res 1995; 21: 199–209.
Auld DS, Kornecook TJ, Bastianetto S, Quirion R . Alzheimer's disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol 2002; 68: 209–45.
Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, et al. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Compar Neurol 1989; 284: 314–35.
Albuquerque EX, Alkondon M, Pereira EF, Castro NG, Schrattenholz A, Barbosa CT, et al. Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther 1997; 280: 1117–36.
Tribollet E, Bertrand D, Marguerat A, Raggenbass M . Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: an autoradiographic study in the rat brain. Neuroscience 2004; 124: 405–20.
Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A . Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-alpha-bungarotoxin. J Neurosci 1985; 5: 1307–15.
Court JA, Lloyd S, Johnson M, Griffiths M, Birdsall NJ, Piggott MA, et al. Nicotinic and muscarinic cholinergic receptor binding in the human hippocampal formation during development and aging. Brain Res Dev Brain Res 1997; 101: 93–105.
Deneris ES, Connolly J, Boulter J, Wada E, Wada K, Swanson LW, et al. Primary structure and expression of beta 2: a novel subunit of neuronal nicotinic acetylcholine receptors. Neuron 1988; 1: 45–54.
Dineley-Miller K, Patrick J . Gene transcripts for the nicotinic acetylcholine receptor subunit, beta4, are distributed in multiple areas of the rat central nervous system. Brain Res Mol Brain Res 1992; 16: 339–44.
Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, et al. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci 1992; 12: 2765–84.
Marks MJ, Pauly JR, Grun EU, Collins AC . ST/b and DBA/2 mice differ in brain alpha-bungarotoxin binding and alpha 7 nicotinic receptor subunit mRNA levels: a quantitative autoradiographic analysis. Brain Res Mol Brain Res 1996; 39: 207–22.
Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW . Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 1993; 13: 596–604.
Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW . The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (alpha 5) in the rat central nervous system. Brain Res 1990; 526: 45–53.
Zarei MM, Radcliffe KA, Chen D, Patrick JW, Dani JA . Distributions of nicotinic acetylcholine receptor alpha7 and beta2 subunits on cultured hippocampal neurons. Neuroscience 1999; 88: 755–64.
Hellstrom-Lindahl E, Mousavi M, Zhang X, Ravid R, Nordberg A . Regional distribution of nicotinic receptor subunit mRNAs in human brain: comparison between Alzheimer and normal brain. Brain Res Mol Brain Res 1999; 66: 94–103.
Graham AJ, Ray MA, Perry EK, Jaros E, Perry RH, Volsen SG, et al. Differential nicotinic acetylcholine receptor subunit expression in the human hippocampus. J Chem Neuroanat 2003; 25: 97–113.
Alkondon M, Pereira EF, Albuquerque EX . Mapping the location of functional nicotinic and gamma-aminobutyric acid A receptors on hippocampal neurons. J Pharmacol Exp Ther 1996; 279: 1491–506.
Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA . Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 1996; 383: 713–6.
Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, et al. Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci 2001; 21: 7993–8003.
Khiroug L, Giniatullin R, Klein RC, Fayuk D, Yakel JL . Functional mapping and Ca2+ regulation of nicotinic acetylcholine receptor channels in rat hippocampal CA1 neurons. J Neurosci 2003; 23: 9024–31.
Yakel JL, Shao Z . Functional and molecular characterization of neuronal nicotinic ACh receptors in rat hippocampal interneurons. Prog Brain Res 2004; 145: 95–107.
Wonnacott S . Presynaptic nicotinic ACh receptors. Trends Neurosci 1997; 20: 92–8.
MacDermott AB, Role LW, Siegelbaum SA . Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci 1999; 22: 443–85.
McKay BE, Placzek AN, Dani JA . Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 2007; 74: 1120–33.
Alkondon M, Rocha ES, Maelicke A, Albuquerque EX . Diversity of nicotinic acetylcholine receptors in rat brain. V. alpha-Bungarotoxin-sensitive nicotinic receptors in olfactory bulb neurons and presynaptic modulation of glutamate release. J Pharmacol Exp Ther 1996; 278: 1460–71.
Role LW, Berg DK . Nicotinic receptors in the development and modulation of CNS synapses. Neuron 1996; 16: 1077–85.
Jones S, Sudweeks S, Yakel JL . Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci 1999; 22: 555–61.
Radcliffe KA, Fisher JL, Gray R, Dani JA . Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann N Y Acad Sci 1999; 868: 591–610.
Ji D, Lape R, Dani JA . Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron 2001; 31: 131–41.
Ge S, Dani JA . Nicotinic acetylcholine receptors at glutamate synapses facilitate long-term depression or potentiation. J Neurosci 2005; 25: 6084–91.
McGehee DS, Role LW . Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol 1995; 57: 521–46.
Dajas-Bailador FA, Heimala K, Wonnacott S . The allosteric potentiation of nicotinic acetylcholine receptors by galantamine is transduced into cellular responses in neurons: Ca2+ signals and neurotransmitter release. Mol Pharmacol 2003; 64: 1217–26.
Rathouz MM, Vijayaraghavan S, Berg DK . Acetylcholine differentially affects intracellular calcium via nicotinic and muscarinic receptors on the same population of neurons. J Biol Chem 1995; 270: 14366–75.
Rathouz MM, Vijayaraghavan S, Berg DK . Elevation of intracellular calcium levels in neurons by nicotinic acetylcholine receptors. Mol Neurobiol 1996; 12: 117–31.
Sharma G, Vijayaraghavan S . Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 2003; 38: 929–39.
Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV . Synaptic potentials mediated via alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci 1998; 18: 8228–35.
Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, et al. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci 1997; 17: 9165–71.
Umbriaco D, Garcia S, Beaulieu C, Descarries L . Relational features of acetylcholine, noradrenaline, serotonin and GABA axon terminals in the stratum radiatum of adult rat hippocampus (CA1). Hippocampus 1995; 5: 605–20.
Papke RL, Bencherif M, Lippiello P . An evaluation of neuronal nicotinic acetylcholine receptor activation by quaternary nitrogen compounds indicates that choline is selective for the alpha 7 subtype. Neurosci Lett 1996; 213: 201–4.
Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX . Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci 1997; 9: 2734–42.
Uteshev VV, Meyer EM, Papke RL . Regulation of neuronal function by choline and 4OH-GTS-21 through alpha 7 nicotinic receptors. J Neurophysiol 2003; 89: 1797–806.
Ji D, Dani JA . Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol 2000; 83: 2682–90.
Alkondon M, Albuquerque EX . Nicotinic acetylcholine receptor alpha7 and alpha4beta2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J Neurophysiol 2001; 86: 3043–55.
Alkondon M, Pereira EF, Albuquerque EX . alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res 1998; 810: 257–63.
Mansvelder HD, McGehee DS . Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 2000; 27: 349–57.
Dani JA, Ji D, Zhou FM . Synaptic plasticity and nicotine addiction. Neuron 2001; 31: 349–52.
Pidoplichko VI, Dani JA . Applying small quantities of multiple compounds to defined locations of in vitro brain slices. J Neurosci Methods 2005; 142: 55–66.
Mansvelder HD, Lodder JC, Sons MS, Kits KS . Dopamine modulates exocytosis independent of Ca2+ entry in melanotropic cells. J Neurophysiol 2002; 87: 793–801.
Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA . Upregulation of surface alpha4beta2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci 1999; 19: 4804–14.
Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA . Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci 2003; 23: 3176–85.
Hasselmo ME . The role of acetylcholine in learning and memory. Curr Opin Neurobiol 2006; 16: 710–5.
Levin ED, McClernon FJ, Rezvani AH . Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006; 184: 523–39.
Levin ED, Kim P, Meray R . Chronic nicotine working and reference memory effects in the 16-arm radial maze: interactions with D1 agonist and antagonist drugs. Psychopharmacology (Berl) 1996; 127: 25–30.
Levin ED, Kaplan S, Boardman A . Acute nicotine interactions with nicotinic and muscarinic antagonists: working and reference memory effects in the 16-arm radial maze. Behav Pharmacol 1997; 8: 236–42.
Levin ED, Bradley A, Addy N, Sigurani N . Hippocampal alpha 7 and alpha 4 beta 2 nicotinic receptors and working memory. Neuroscience 2002; 109: 757–65.
Nott A, Levin ED . Dorsal hippocampal alpha7 and alpha4beta2 nicotinic receptors and memory. Brain Res 2006; 1081: 72–8.
Levin ED, Simon BB . Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology (Berl) 1998; 138: 217–30.
Levin ED, Torry D . Acute and chronic nicotine effects on working memory in aged rats. Psychopharmacology (Berl) 1996; 123: 88–97.
Socci DJ, Sanberg PR, Arendash GW . Nicotine enhances Morris water maze performance of young and aged rats. Neurobiol Aging 1995; 16: 857–60.
Song C, Murray TA, Kimura R, Wakui M, Ellsworth K, Javedan SP, et al. Role of alpha7-nicotinic acetylcholine receptors in tetanic stimulation-induced gamma oscillations in rat hippocampal slices. Neuropharmacology 2005; 48: 869–80.
Malenka RC, Bear MF . LTP and LTD: an embarrassment of riches. Neuron 2004; 44: 5–21.
Martin SJ, Grimwood PD, Morris RG . Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 2000; 23: 649–711.
Brown RE, Milner PM . The legacy of Donald O. Hebb: more than the Hebb synapse. Nat Rev 2003; 4: 1013–9.
Whitlock JR, Heynen AJ, Shuler MG, Bear MF . Learning induces long-term potentiation in the hippocampus. Science 2006; 313: 1093–7.
Yamazaki Y, Jia Y, Hamaue N, Sumikawa K . Nicotine-induced switch in the nicotinic cholinergic mechanisms of facilitation of long-term potentiation induction. Eur J Neurosci 2005; 22: 845–60.
Lagostena L, Trocme-Thibierge C, Morain P, Cherubini E . The partial alpha7 nicotine acetylcholine receptor agonist S 24795 enhances long-term potentiation at CA3-CA1 synapses in the adult mouse hippocampus. Neuropharmacology 2008; 54: 676–85.
Alkondon M, Pereira EF, Almeida LE, Randall WR, Albuquerque EX . Nicotine at concentrations found in cigarette smokers activates and desensitizes nicotinic acetylcholine receptors in CA1 interneurons of rat hippocampus. Neuropharmacology 2000; 39: 2726–39.
Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T . Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. Eur J Neurosci 2000; 12: 3741–7.
Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci 2007; 27: 8202–18.
Kauer JA, Malenka RC . Synaptic plasticity and addiction. Nat Rev 2007; 8: 844–58.
Saal D, Dong Y, Bonci A, Malenka RC . Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 2003; 37: 577–82.
See RE . Neural substrates of conditioned-cued relapse to drug-seeking behavior. Pharmacol Biochem Behav 2002; 71: 517–29.
Dani JA, Harris RA . Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci 2005; 8: 1465–70.
Slemmer JE, Martin BR, Damaj MI . Bupropion is a nicotinic antagonist. J Pharmacol Exp Ther 2000; 295: 321–7.
Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, et al. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem 2005; 48: 3474–7.
Mihalak KB, Carroll FI, Luetje CW . Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol 2006; 70: 801–5.
Frotscher M, Leranth C . Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. J Compar Neurol 1985; 239: 237–46.
Acknowledgements
Dr John Anthony DANI is supported by multiple grants from the National Institutes of Health (NS21229, DA09411); Tao A ZHANG is supported by a Philip Morris postdoctoral fellowship, and Andon Nicholas PLACZEK is supported by a National Institute of Drug Abuse postdoctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Placzek, A., Zhang, T. & Dani, J. Nicotinic mechanisms influencing synaptic plasticity in the hippocampus. Acta Pharmacol Sin 30, 752–760 (2009). https://doi.org/10.1038/aps.2009.39
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.39
Keywords
This article is cited by
-
Regulation of habenular G-protein gamma 8 on learning and memory via modulation of the central acetylcholine system
Molecular Psychiatry (2021)
-
Memory enhancing effects of nicotine, cocaine, and their conditioned stimuli; effects of beta-adrenergic and dopamine D2 receptor antagonists
Psychopharmacology (2021)
-
Compromised neuroplasticity in cigarette smokers under nicotine withdrawal is restituted by the nicotinic α4β2-receptor partial agonist varenicline
Scientific Reports (2017)
-
TRPV1 regulates excitatory innervation of OLM neurons in the hippocampus
Nature Communications (2017)
-
Differential modulation of GABAA and NMDA receptors by α7-nicotinic receptor desensitization in cultured rat hippocampal neurons
Acta Pharmacologica Sinica (2016)
